Vispārība
Osteogenesis imperfecta ir iedzimta ģenētiska slimība, kas nav saistīta ar dzimumu un ir atbildīga par noteiktu kaulu trauslumu un izteiktu lūzumu tendenci.
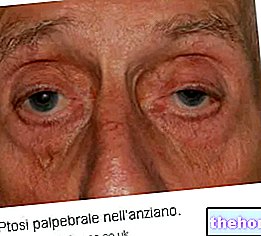
Osteogenesis imperfecta simptomi ir daudz; parasti tie sastāv no: kaulu vājināšanās, augsta tendence uz kaulu lūzumiem, zilu, pelēku vai purpursarkanu acu sklēru klātbūtne, kaulu deformācijas vai citas skeleta izmaiņas, trīsstūrveida seja, zobu trauslums utt. .
Kopumā, lai pareizi diagnosticētu osteogenesis imperfecta, ir būtiski šādi faktori: fiziskā pārbaude, medicīniskā vēsture, medicīniskās attēlveidošanas testi, I tipa kolagēna novērtēšanas tests un ģenētiskais tests.
Diemžēl pašlaik vienīgās ārstēšanas iespējas pacientiem ar osteogenesis imperfecta ir simptomātiskas. Attiecīgā slimība patiesībā ir neārstējama.
Kas ir osteogenesis imperfecta?
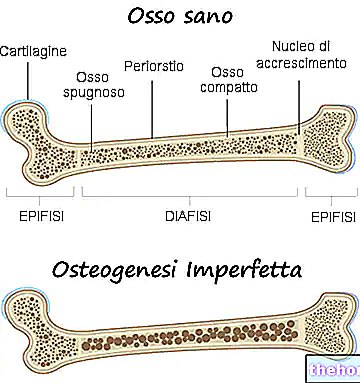
Osteogenesis imperfecta ir ģenētiska slimība, kuras dēļ skartās personas kauli kļūst vājāki un vairāk pakļauti lūzumiem.
Patiesībā ar terminu osteogenesis imperfecta ārsti atsaucas uz neviendabīgu ģenētisko slimību grupu, kurai raksturīga noteikta kaulu trausluma pakāpe. Tāpēc pastāv vairākas osteogenesis imperfecta formas (vai veidi), dažas ir daudz smagākas nekā citas.
Tā ir iedzimta slimība
Cilvēkiem, kurus tā skārusi, osteogenesis imperfecta ir slimība, kas pastāv jau kopš dzimšanas, tāpēc to visumā var definēt kā iedzimtu slimību.
Vai tas ir saistīts ar seksu?
Osteogenesis imperfecta nav ar dzimumu saistīta ģenētiska slimība, piemēram, hemofilija vai Klinefeltera sindroms.
EPIDEMILOĢIJA
Saskaņā ar dažiem statistikas pētījumiem, osteogenesis imperfecta sastopamība būtu vienāda ar vienu gadījumu ik pēc 15 000–20 000 jaundzimušo. Tas nozīmē, ka katriem 15 000–20 000 jaundzimušajiem ir kāds, kuru skārusi nepilnīga osteogeneze.
Citi statistikas pētījumi arī ir parādījuši, ka osteogenesis imperfecta vienlīdz ietekmē vīriešus un sievietes, un ka tā nedod priekšroku konkrētai populācijai vai etniskajai grupai.
Dzīves ilgums ir ļoti mainīgs parametrs, kas ir atkarīgs no nepilnīgas osteogenezes formas.
Cēloņi
Osteogenesis imperfecta gandrīz vienmēr rodas kvalitatīvu un kvantitatīvu I tipa kolagēna ražošanas izmaiņu rezultātā.
I tipa kolagēns ir būtisks, lai stiprinātu kaulus un uzturētu veselus saistaudus, kas veido skrimšļus, cīpslas, ādu, acu sklēru utt.
Tāpēc izmaiņas I tipa kolagēna ražošanā ietekmē kaulu izturību un cilvēka organismā esošo saistaudu veselību.
Kas maina kolagēna ražošanu?
Ģenētiska slimība ir stāvoklis, kas rodas viena vai vairāku šūnu DNS veidojošu gēnu mutācijas dēļ.
Osteogenesis imperfecta gadījumā tā cēloņi gandrīz vienmēr ir meklējami viena vai abu gēnu COL1A1 (atrodas 17. hromosomā) un COL1A2 (atrodas 7. hromosomā) mutācijā.
Normālos apstākļos COL1A1 un COL1A2 regulē normālu I tipa kolagēna ražošanu; mutāciju klātbūtnē viņu lādē viņi neveic savu regulējošo funkciju.
Svarīgi: kādi citi gēni, ja tie ir mutēti, izraisa osteogenesis imperfecta?
Papildus COL1A1 un COL1A2 mutācijām IFITM5, SERPINF1, CRTAP un LEPRE1 gēnu mutācijas ir potenciāli osteogenesis imperfecta cēloņi.
Iepriekš minētie gēni aptver funkcijas, kas atšķiras no COL1A1 un COL1A2 - tādēļ tie nekontrolē I tipa kolagēna ražošanu -, taču tiem joprojām ir "ietekme uz cilvēka skeleta kaulu izturību un pretestību.
KĀDA ĢENĒTISKĀ SLIMĪBA TAS IR?
Osteogenesis imperfecta ir autosomāli ģenētiska slimība.
Termins autosomāls, kas saistīts ar ģenētisku slimību, norāda, ka attiecīgais stāvoklis ir saistīts ar ģenētiskām mutācijām, kuru pamatā ir autosomālās un ne-dzimuma hromosomas.
Lasītājiem tiek atgādināts, ka cilvēkam ir hromosomu kopums, kas sastāv no 23 pāriem kopējo hromosomu, kurā 22 pāri ir autosomāli un tikai viens pāris ir seksuāla tipa. Seksuālā tipa hromosomu pāris ietekmē dzimumu. individuāls.
Osteogenesis imperfecta pēc mutācijām COL1A1, COL1A2 un IFITM5 piemīt visas autosomāli dominējošās slimības pazīmes. Ja tas ir saistīts ar SERPINF1, CRTAP un LEPRE1 gēnu mutācijām, tam piemīt autosomāli recesīvas slimības pazīmes.
VEIDI
Pašlaik ārsti uzskata, ka pastāv 8 osteogenesis imperfecta veidi (vai formas). Lai atšķirtu dažādus veidus, viņi nolēma izmantot romiešu numerāciju, precīzāk sakot, pirmos astoņus romiešu ciparus.
Zemāk esošajā tabulā ir parādītas 8 nepilnīgās osteogenesis formas, tās izraisošās mutācijas un citas pazīmes.
Puisis
Mutēts gēns
Ģenētiskās slimības veids
THE
COL1A1
Autosomāli dominējošs
II
COL1A1 un COL1A2
Autosomāli dominējošs
III
COL1A1 un COL1A2
Autosomāli dominējošs
IV
COL1A1 un COL1A2
Autosomāli dominējošs
V.
IFITM5
Autosomāli dominējošs
TU
SERPINF1
Autosomāli recesīvs
VII
CRTAP
Autosomāli recesīvs
VIII
HARE 1
Autosomāli recesīvs
* NB! Acīmredzot COL1A1 un COL1A2 mutācijas, kas izraisa pirmās četras osteogenesis imperfecta formas, ir ģenētiskas izmaiņas ar nedaudz atšķirīgām īpašībām. Pretējā gadījumā nebūtu jēgas atšķirt vienu no otra.
Simptomi, pazīmes un komplikācijas
Visu veidu osteogenesis imperfecta ir atbildīgi par kaulu vājināšanos, tā ka slimības skartajai personai ir īpaša nosliece uz lūzumiem. Kaulu vājināšanās pakāpe atšķiras atkarībā no formas; dažiem no tiem šī vājināšanās ir lielāka nekā citiem.
To sakot, jānorāda, ka katrai osteogenesis imperfecta formai ir savs simptomātisks attēls, kas dažiem var atgādināt citu formu simptomatoloģisko ainu.
IESPĒJAMIE SIMPTOMI UN ZĪMES
Iespējamie osteogenesis imperfecta simptomi un pazīmes ir šādas:
- Kaulu malformāciju klātbūtne;
- Īsa un maza ķermeņa klātbūtne (paredzēta kā stumbrs);
- Locītavu problēmas (piemēram, vaļīgas locītavas);
- Muskuļu vājums;
- Zila, violeta vai pelēka acu sklēra;
- Trīsstūrveida seja;
- Mucas lāde;
- Mugurkaula morfoloģiskās anomālijas;
- Zobu trauslums;
- Dzirdes samazināšanās vai pilnīga zaudēšana;
- Elpošanas problēmas
- Problēmas, kas saistītas ar 1. tipa kolagēna neesamību vai trūkumu.
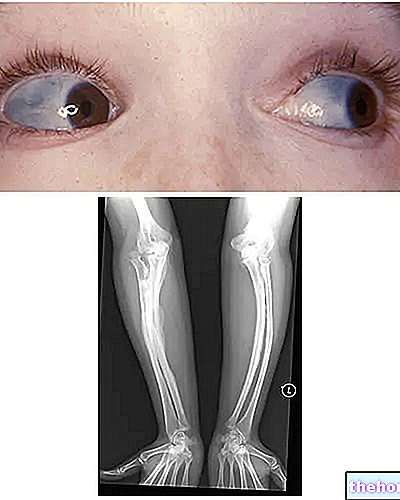
Osteogenesis Imperfecta: ņemiet vērā sklēru zilo krāsu un kaulu deformācijas, kas raksturo slimību. No wikipedia.org
KAS IR NOPIETNĪGĀKĀ NEVEIKTĀS OSTEOGENĒZES FORMA?
Ārsti klasificē dažādu osteogenesis imperfecta veidu simptomātisko smagumu skalā no 3 grādiem, kas ir: viegla pakāpe, mērena pakāpe un smaga pakāpe.
Tikai viena forma pieder "vieglas pakāpes" kategorijai: "I tipa osteogenesis imperfecta"; 4 osteogenesis imperfecta formas pieder pie "vidējas pakāpes" kategorijas: IV, V un VI; visbeidzot, "smagas pakāpes" kategorijai pieder 3. veidlapas: II, III, VII un VIII.
I TIPS: FUNKCIJAS
Visizplatītākajai un vismazāk smagajai I tipa osteogenesis imperfecta formai ir šādas īpašības:
- Tas izraisa lūzumus, īpaši pirms pubertātes;
- Tas "gandrīz neietekmē augumu, tāpēc pacienti parasti ir normālā augumā";
- Izraisa locītavu problēmas un muskuļu vājumu
- Tas ir atbildīgs par zilu, violetu vai pelēku sklēru;
- Tas ir trīsstūrveida sejas un mugurkaula anomāliju cēlonis;
- Tas gandrīz nekad neizraisa kaulu deformācijas. Ja tas viņus provocē, tie ir minimāli;
- Tas var izraisīt zobu trauslumu un / vai dzirdes zudumu (pēdējais parasti rodas pieaugušā vecumā);
- Tas ir saistīts ar I tipa kolagēna klātbūtni, kura kvalitāte ir normāla, bet daudzums ir neparasts (tas ir sliktāks nekā parasti).
II TIPS: ĪPAŠĪBAS
II tipa osteogenesis imperfecta raksturo:
- Nāves cēlonis dzimšanas brīdī vai neilgi pēc tam. Elpošanas problēmas gandrīz vienmēr izraisa nāvi;
- Ievērojama kaulu trausluma un smagu kaulu deformāciju klātbūtne;
- Īss augums un nepietiekami attīstītas plaušas
- Zila, violeta vai pelēka sklēra;
- I tipa kolagēna kvantitatīvo un kvalitatīvo anomāliju klātbūtne.
III VEIDS: FUNKCIJAS
III tipa osteogenesis imperfecta ir šādas īpašības:
- Lai gan tas ir ļoti nopietni, jaundzimušo periodā tas bieži neizraisa nāvi;
- Tas ir saistīts ar "augstu kaulu trauslumu;
- Tas ir atbildīgs par mazu augumu, locītavu problēmām, muskuļu vājumu (īpaši kājās un rokās), mucas krūtīm, trīsstūrveida seju un patoloģisku mugurkaula izliekumu;
- Tas ir saistīts ar zilu, violetu vai pelēku sklēru;
- Tas var izraisīt elpošanas problēmas, zobu trauslumu un dzirdes zudumu;
- Tas bieži ir atbildīgs par kaulu deformācijām;
- Tas ir saistīts ar I tipa kolagēna kvalitatīvām un kvantitatīvām novirzēm.
IV TIPS: ĪPAŠĪBAS
IV tipa osteoģenēzi raksturo:
- Kaulu trausluma pakāpe starp II un III formu un I formu;
- Īsāks par vidējo augumu;
- Zila, violeta vai pelēka sklēra;
- Vieglas / vidēji smagas formas kaulu deformācijas, nelielas mugurkaula un cilindra krūškurvja novirzes;
- Trīsstūrveida seja;
- Iespējama zobu trausluma un dzirdes zuduma klātbūtne;
- I tipa kolagēna patoloģiju klātbūtne.
V TIPS: FUNKCIJAS
V tipa osteogenesis imperfecta dažos veidos atgādina IV tipa osteogenesis imperfecta. Tomēr tam ir dažas īpatnības, proti:
- Normālas krāsas sklera;
- Zobu trausluma trūkums;
- Patoloģisku kaulu kaulu veidošanās kaulu lūzumu dzīšanas procesā;
- Starpslāņu membrānas, kas atrodas starp rādiusu un elkoņa kauli, pārkaļķošanās. Tas pasliktina apakšdelma kustīgumu.
VI VEIDS: FUNKCIJAS
Arī VI tipa osteogenesis imperfecta ir līdzīgs IV formai. Lai to atšķirtu no pēdējās, ir dažas īpatnības, tostarp augsts sārmainās fosfatāzes līmenis asinīs un dažos kaulos esošas lamelītes (kaulainas), kas līdzīgas zivju muguriņām.
VII TIPS: ĪPAŠĪBAS
Simptomātiski VII tipa osteogenesis imperfecta dažos apstākļos var līdzināties IV tipam un citos - II tipam.
Šīs nopietnās patoloģiskās formas īpatnības ir šādas:
- Īss augums;
- Ārkārtīgi īsa pleca kaula (rokas kaula) un augšstilba kaula (augšstilba kaula) klātbūtne;
- Bieža gūžas deformācija, kas pazīstama kā coxa vara.
VIII TIPS: RAKSTUROJUMS
VIII tipa osteogenesis imperfecta ļoti atgādina II un III formu.
Starp tās īpatnībām izceļas: smags augšanas deficīts, smaga skeleta hipomineralizācija un prolil-3-hidroksilāzes enzīma neesamība (vai nepietiekama klātbūtne).
Diagnoze
Parasti diagnostikas process, kuram pakļauti pacienti ar aizdomām par osteogenesis imperfecta formu, sākas ar rūpīgu fizisku pārbaudi un rūpīgu slimības vēsturi; tad tas turpinās, "analizējot pacienta ģimenes vēsturi un veicot virkni diagnostisko attēlveidošanas testu (rentgena, CT skenēšana utt.); visbeidzot, tas beidzas ar I tipa kolagēna kvantitatīvu un kvalitatīvu novērtējumu un ģenētiskais tests.
Mūsdienās pastāv iespēja diagnosticēt osteogenesis imperfecta pat pirmsdzemdību fāzē, pakļaujot grūtnieci ultraskaņai.
OBJEKTĪVĀS PĀRBAUDES UN VĒSTURES SVARĪGUMS
Ārsts eksperts osteogenesis imperfecta ļoti bieži spēj diagnosticēt iepriekš minēto slimību pat tikai, veicot fizisku pārbaudi un anamnēzi. Tas nozīmē, ka šiem diagnostikas testiem nav mazsvarīgas nozīmes.
I TIPA KOLAGENA RAŽOŠANAS NOVĒRTĒŠANA
Parasti I tipa kolagēna kvalitatīvais un kvantitatīvais novērtējums ir ļoti uzticams tests, jo, kā minēts, vairumam osteogenesis imperfecta gadījumu raksturīgas mutācijas gēnos, kas kontrolē 1. tipa kolagēna ražošanu.
Lai novērtētu indivīda šūnu līmenī esošā I tipa kolagēna daudzumu un kvalitāti, ārsti var paļauties uz ādas biopsiju vai īpašu asins analīzi.
Abi šie novērtēšanas testi ir diezgan sarežģīti, un pacientam (vai viņa vecākiem), iespējams, būs jāgaida vairākas nedēļas, lai uzzinātu rezultātus.
ĢENĒTISKAIS TESTS
Izmantojot ģenētisko testu, kas pārbauda visu pārbaudāmās personas DNS, ārsti var galīgi noteikt esošās ģenētiskās mutācijas īpašības.
Parasti ģenētiskā testa veikšana uz visu šūnu DNS ir paredzēta, ja I tipa kolagēna īpašību novērtējums nav devis vēlamos rezultātus vai ja tā nav mutācija COL1A1 vai COL1A2, kas izraisa "osteogenesis imperfecta".
PRENATĀLĀ DIAGNOZE
Pirmsdzemdību ultraskaņa ir ļoti noderīga, lai identificētu II un III tipa osteogenesis imperfecta.
Terapija
Pašlaik nav īpašas zāles osteogenesis imperfecta ārstēšanai. Citiem vārdiem sakot, cilvēkiem ar osteogenesis imperfecta ir lemts dzīvot kopā ar iepriekš minēto stāvokli līdz nāvei, kas bieži vien ir saistīta ar pašas slimības sekām.
Specifiskas terapijas trūkums neizslēdz citu ārstēšanas veidu esamību. Patiesībā starp pacienta ar osteogenesis imperfecta terapeitiskajām iespējām ir iekļautas dažādas simptomātiskas terapijas; ar simptomātisku terapiju mēs saprotam ārstēšanu, kas spēj mazināt simptomus, palēnināt slimības gaitu un novērst (vai vismaz atlikt) visnopietnākās sekas.
IESPĒJAMĀS SIMPTOMĀTISKĀS APSTRĀDES
Iespējamo simptomātisko osteogenesis imperfecta ārstēšanas metožu sarakstā izceļas:
- Nagu ķirurģiska ievietošana garāko kaulu iekšpusē (NB: visbiežāk pakļauti lūzumiem), kas nodrošina lielāku izturību pret lūzumiem un deformācijām. Šo operāciju sauc rodding intramedulāra;
- Lūzumu un / vai kaulu deformāciju konservatīva vai ķirurģiska ārstēšana;
- Zobu aprūpe, lai aizsargātu zobu veselību;
- Sāpju mazinošas terapijas ļoti sāpīgu vairāku lūzumu gadījumā;
- Fizioterapija, muskuļu pagarināšanai un stiprināšanai.Elastīgs un tonizējošs muskuļu aparāts ļauj novērst kritienus, kas varētu izraisīt dažādus kaulu lūzumus;
- Pārvietošanās palīglīdzekļu izmantošana, ieskaitot ratiņkrēslus, breketes, kruķus utt.
KUSTĪBAS IEGUVUMI
Personām ar osteogenesis imperfecta ārsti iesaka pastāvīgi praktizēt fiziskos vingrinājumus un kustību kopumā, jo abas šīs aktivitātes veicina skeleta un muskuļu sistēmas nostiprināšanos.
Starp ieteicamajiem sporta veidiem ir: peldēšana, jo tā ir “fiziska aktivitāte ar zemu ietekmi uz skeleta sistēmu”, un pastaigas.
Ieguvumi no veselīga dzīvesveida
Veselīga dzīvesveida vadīšana, izvairīšanās no smēķēšanas, pārmērīga alkohola lietošana, pārāk daudz un slikta ēšana utt., Ir vairāk nekā diskrēts ieguvums veselībai pacientiem ar osteogenesis imperfecta, jo tas palēnina slimības progresēšanu un samazina kaulu trauslumu.
SIMPTOMĀTISKĀ ĀRSTĒŠANA EKSPERIMENTĀCIJAS FĀZĒ
Pašlaik ārsti un pētnieki novērtē dažu simptomātisku ārstēšanas metožu efektivitāti, ieskaitot augšanas hormona terapiju un bifosfonātu saturošu intravenozu un perorālu terapiju.
Šobrīd rezultāti, ko sniedz iepriekš minētās izmeklēšanas procedūras, liecina par labu visai medicīnas sabiedrībai.
Prognoze
Osteogenesis imperfecta ir slimība ar negatīvu prognozi, jo tā ir neārstējama, krasi pasliktina dzīves kvalitāti un dažos gadījumos izraisa skartā subjekta priekšlaicīgu nāvi.
Tomēr jāatzīmē, ka, arī pateicoties mūsdienu simptomātiskai ārstēšanai, daudzi cilvēki ar vieglu osteogenesis imperfecta formu spēj dzīvot patīkami un apmierinoši.
Profilakse
Diemžēl pašlaik nav profilakses līdzekļa pret osteogenesis imperfecta.





-cos-cause-e-terapia.jpg)