Aktīvās sastāvdaļas: Erlotinibs
Tarceva 25 mg apvalkotās tabletes
Tarceva 100 mg apvalkotās tabletes
Tarceva 150 mg apvalkotās tabletes
Kāpēc lieto Tarceva? Kam tas paredzēts?
Tarceva satur aktīvo vielu erlotinibu. Tarceva ir zāles, ko lieto vēža ārstēšanai, un tās bloķē proteīna, ko sauc par epidermas augšanas faktora receptoru (EGFR), darbību, kas ir iesaistīts vēža šūnu augšanā un izplatībā.
Tarceva ir indicēts pieaugušajiem. Šīs zāles var parakstīt Jums, ja Jums ir progresējošs nesīkšūnu plaušu vēzis. To var parakstīt kā sākotnējo terapiju vai terapiju, ja pēc sākotnējās ķīmijterapijas slimība lielākoties nemainās, ja vien vēža šūnām ir specifiskas EGFR mutācijas. To var parakstīt arī tad, ja iepriekšējā ķīmijterapija nav spējusi apturēt vēzi . slimība.
Ja Jums ir metastātisks aizkuņģa dziedzera vēzis, šīs zāles Jums var parakstīt arī kopā ar citu ārstēšanu, ko sauc par gemcitabīnu.
Kontrindikācijas Kad Tarceva nedrīkst lietot
Nelietojiet Tarceva
- ja Jums ir alerģija pret erlotinibu vai kādu citu (6. punktā minēto) šo zāļu sastāvdaļu.
Piesardzība lietošanā Kas jāzina pirms Tarceva lietošanas
Brīdinājumi un piesardzība lietošanā:
- ja lietojat citas zāles, kas var palielināt vai samazināt erlotiniba daudzumu asinīs vai ietekmēt tā efektivitāti (piemēram, pretsēnīšu līdzekļus, piemēram, ketokonazolu, proteāzes inhibitorus, eritromicīnu, klaritromicīnu, fenitoīnu, karbamazepīnu, barbiturātus, rifampicīnu, ciprofloksacīnu, omeprazolu, ranitidīnu, asinszāli vai proteasomu inhibitorus), jautājiet savam ārstam. Dažos gadījumos šīs zāles var samazināt Tarceva efektivitāti vai palielināt blakusparādības, un, ja tā, tad ārstam, iespējams, būs jāpielāgo terapija.Ārsts var noorganizēt, ka Jūs šīs zāles nelietojat ārstēšanas laikā ar Tarceva.
- ja lietojat antikoagulantus (zāles, kas palīdz novērst trombozi vai asins recēšanu, piemēram, varfarīnu), Tarceva var palielināt tieksmi uz asiņošanu. Konsultējieties ar savu ārstu, kuram jums būs jāuzrauga, periodiski izrakstot dažas asins analīzes.
- ja lietojat statīnus (zāles holesterīna līmeņa pazemināšanai asinīs), Tarceva var palielināt ar statīniem saistītu muskuļu problēmu risku, kas retos gadījumos var izraisīt smagu muskuļu sabrukumu (rabdomiolīzi), izraisot nieru bojājumus. Konsultējieties ar savu ārstu.
- ja lietojat kontaktlēcas un / vai kādreiz ir bijušas acu problēmas, piemēram, smagas sausas acis, acs priekšējās daļas (radzenes) iekaisums vai čūlas, kas skārušas acs priekšpusi, konsultējieties ar ārstu. Skatīt arī apakšpunktā "Citas zāles un Tarceva".
Jums jāinformē ārsts:
- ja Jums ir “pēkšņas elpošanas grūtības, kas saistītas ar klepu vai drudzi, jo ārstam var būt nepieciešams izrakstīt citas zāles un pārtraukt Tarceva lietošanu;
- ja Jums ir caureja, jo ārstam var būt nepieciešams izrakstīt pretcaurejas līdzekļus (piemēram, loperamīdu);
- nekavējoties, ja Jums ir smaga vai ilgstoša caureja, slikta dūša, apetītes zudums vai vemšana, jo ārstam var būt nepieciešams pārtraukt Tarceva terapiju un Jums nepieciešama ārstēšana slimnīcā.
- ja Jums ir stipras sāpes vēderā, smagas ādas reakcijas, piemēram, pūslīši vai lobīšanās.Ārsts var uzskatīt par nepieciešamu pārtraukt vai pārtraukt ārstēšanu.
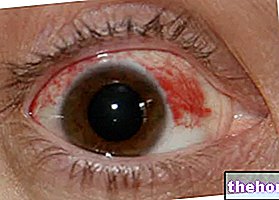
- ja Jums rodas akūts sarkans vai pastiprinās acu apsārtums, ko pavada sāpes, pastiprināta asarošana, neskaidra redze un / vai jutība pret gaismu, nekavējoties konsultējieties ar ārstu vai medmāsu, jo Jums var būt nepieciešama steidzama ārstēšana (skatīt Iespējamās blakusparādības).
- ja lietojat arī statīnu un rodas neizskaidrojamas muskuļu sāpes, jutīgums, vājums vai krampji. Ārsts var uzskatīt par nepieciešamu pārtraukt vai pārtraukt ārstēšanu.
Skatīt arī 4. sadaļu "Iespējamās blakusparādības".
Aknu un nieru slimības
Nav zināms, vai Tarceva iedarbība mainās, ja aknas vai nieres nedarbojas normāli. Ja Jums ir smaga aknu vai nieru slimība, ārstēšana ar šīm zālēm nav ieteicama.
Glikuronācijas traucējumi, piemēram, Gilberta sindroms
Ja Jums ir glikuronidācijas traucējumi, piemēram, Gilberta sindroms, ārstam pret jums jārīkojas piesardzīgi.
Dūmi
Ja lietojat Tarceva, jums jāpārtrauc smēķēšana, jo smēķēšana var samazināt zāļu daudzumu asinīs.
Bērni un pusaudži
Tarceva nav pētīts pacientiem līdz 18 gadu vecumam. Ārstēšana ar šīm zālēm nav ieteicama bērniem un pusaudžiem.
Mijiedarbība Kādas zāles vai pārtikas produkti var mainīt Tarceva iedarbību
Citas zāles un Tarceva
Pastāstiet ārstam vai farmaceitam par visām zālēm, kuras lietojat, pēdējā laikā esat lietojis vai varētu lietot.
Tarceva kopā ar uzturu un dzērienu
Nelietojiet Tarceva kopā ar ēdienu. Skatīt arī 3. punktu "Kā lietot Tarceva"
Brīdinājumi Ir svarīgi zināt, ka:
Grūtniecība un zīdīšanas periods
Izvairieties no grūtniecības Tarceva terapijas laikā. Ja domājat, ka varētu iestāties grūtniecība, terapijas laikā un vismaz 2 nedēļas pēc pēdējās tabletes lietošanas izmantojiet atbilstošu kontracepcijas līdzekli. Ja Tarceva lietošanas laikā iestājas grūtniecība, nekavējoties pastāstiet ārstam, kurš izlems, vai turpināt ārstēšanu. Ja esat grūtniece, domājat, ka Jums varētu būt grūtniecība vai plānojat grūtniecību, vai ja barojat bērnu ar krūti, pirms šo zāļu lietošanas konsultējieties ar ārstu vai farmaceitu.
Transportlīdzekļu vadīšana un mehānismu apkalpošana
Nav veikti pētījumi par iespējamo Tarceva ietekmi uz spēju vadīt transportlīdzekļus un apkalpot mehānismus, taču ir maz ticams, ka ārstēšana mainīs šo spēju.
Paaugstināta jutība
Tarceva satur cukuru, ko sauc par laktozes monohidrātu. Ja ārsts ir teicis, ka Jums ir kāda cukura nepanesība, pirms Tarceva lietošanas konsultējieties ar ārstu.
Deva, lietošanas veids un laiks Kā lietot Tarceva: Devas
Vienmēr lietojiet šīs zāles tieši tā, kā ārsts Jums stāstījis. Ja rodas šaubas, konsultējieties ar ārstu vai farmaceitu.
Tablete jālieto vismaz vienu stundu pirms ēšanas vai divas stundas pēc ēšanas.
Parastā deva ir viena Tarceva 150 mg tablete dienā, ja Jums ir nesīkšūnu plaušu vēzis. Parastā deva ir viena Tarceva 100 mg tablete dienā, ja Jums ir metastātisks aizkuņģa dziedzera vēzis. Tarceva lieto kombinācijā ar gemcitabīnu.
Ārsts var mainīt devu par 50 mg vienlaikus. Dažādām dozēšanas shēmām Tarceva ir pieejams stiprumos 25 mg, 100 mg vai 150 mg.
Pārdozēšana Ko darīt, ja esat lietojis pārāk daudz Tarceva
Ja esat lietojis Tarceva vairāk nekā noteikts
Nekavējoties sazinieties ar ārstu vai farmaceitu. Var gadīties, ka blakusparādības pasliktinās un ārsts liks Jums pārtraukt to lietot.
Ja esat aizmirsis lietot Tarceva
Ja esat izlaidis vienu vai vairākas Tarceva devas, pēc iespējas ātrāk sazinieties ar ārstu vai farmaceitu. Nelietojiet dubultu devu, lai aizvietotu aizmirsto devu.
Ja pārtraucat lietot Tarceva
Ir svarīgi turpināt lietot Tarceva katru dienu tik ilgi, cik noteicis ārsts.
Ja jums ir kādi jautājumi par šo zāļu lietošanu, jautājiet savam ārstam vai farmaceitam.
Blakusparādības Kādas ir Tarceva blakusparādības?
Tāpat kā citas zāles, šīs zāles var izraisīt blakusparādības, kaut arī ne visiem tās izpaužas.
Ja Jums rodas kāda no zemāk uzskaitītajām blakusparādībām, lūdzu, pēc iespējas ātrāk sazinieties ar savu ārstu. Dažos gadījumos ārstam var būt nepieciešams samazināt Tarceva devu vai pārtraukt terapiju.
- Caureja un vemšana (ļoti bieži, var skart vairāk nekā 1 no 10 pacientiem). Pastāvīga un smaga caureja var izraisīt kālija līmeņa pazemināšanos asinīs un nieru mazspēju, īpaši, ja Jūs vienlaikus tiekat ārstēts ar citām ķīmijterapijas zālēm. Smagas vai ilgstošas caurejas gadījumā nekavējoties sazinieties ar savu ārstu, kurš var izlemt ārstēties slimnīcā.
- Acu kairinājums konjunktivīta / keratokonjunktivīta dēļ (ļoti bieži, var skart vairāk nekā 1 no 10 pacientiem) un keratīts (bieži, var skart līdz 1 no 10 pacientiem).
- Plaušu iekaisuma forma, ko sauc par intersticiālu plaušu slimību (retāk Eiropas pacientiem, bieži japāņu pacientiem: var skart līdz 1 no 100 pacientiem Eiropā un līdz 1 no 10 Japānā). Šī slimība var būt saistīta arī ar jūsu veselības stāvokļa dabisko progresēšanu un dažos gadījumos var būt letāla. Ja Jums rodas tādi simptomi kā "pēkšņas elpošanas grūtības, kas saistītas ar klepu vai drudzi, nekavējoties sazinieties ar savu ārstu, jo Jums var būt šī slimība. Ārsts var nolemt neatgriezeniski pārtraukt terapiju ar Tarceva".
- Novēroti kuņģa -zarnu trakta perforācijas gadījumi (retāk, var rasties līdz 1 no 100 pacientiem). Pastāstiet ārstam, ja Jums rodas stipras sāpes vēderā, kā arī pastāstiet ārstam, ja Jums agrāk ir bijusi peptiska čūla vai divertikulāra slimība, jo tas var palielināt perforācijas risku.
- Retos gadījumos ir novērota aknu mazspēja (reti, var rasties līdz 1 no 1000 pacientiem). Ja Jūsu asins analīzes liecina par nopietnām aknu darbības izmaiņām, ārsts var izlemt pārtraukt terapiju.
Ļoti bieži sastopamas blakusparādības (var skart vairāk nekā 1 no 10 pacientiem):
- Ādas izsitumi, kas var attīstīties vai pasliktināties saulē pakļautajās vietās. Ja esat pakļauts saulei, var būt ieteicams lietot aizsargapģērbu un / vai sauļošanās līdzekļus (piemēram, pamatojoties uz minerālvielām)
- Infekcija
- Apetītes zudums, svara zudums
- Depresija
- Galvassāpes, izmaiņas ādas sajūtā vai nejutīgums ekstremitātēs
- Apgrūtināta elpošana, klepus
- Slikta dūša
- Mutes kairinājumi
- Sāpes vēderā, gremošanas traucējumi un meteorisms
- Izmaiņas asins analīzēs, kas saistītas ar aknu darbību
- Niezoša, sausa āda un matu izkrišana
- Nogurums, drudzis, drebuļi
Biežas blakusparādības (var skart līdz 1 no 10 pacientiem):
- Deguna asiņošana
- Asiņošana no kuņģa vai zarnām
- Iekaisuma reakcijas ap nagiem
- Matu folikulu infekcija
- Pūtītes
- Sadalīta āda (ādas plaisas)
- Pavājināta nieru darbība (ja to lieto ārpus apstiprinātajām indikācijām kombinācijā ar ķīmijterapiju)
Retākas blakusparādības (var skart līdz 1 no 100 cilvēkiem)
- skropstu izmaiņas
- pārmērīgi mati uz sejas un ķermeņa ar vīriešu rakstura sadalījumu
- uzacu izmaiņas
- trausli un lobīti nagi
Retas blakusparādības (var skart līdz 1 no 1000 pacientiem):
- plaukstu vai pēdu apsārtums vai sāpes (plaukstas-plantāra eritrodizestēzijas sindroms)
Ļoti retas blakusparādības (var skart līdz 1 no 10 000 pacientiem)
- Radzenes čūlas vai perforācijas gadījumi
- Smagas ādas reakcijas, piemēram, pūslīšu veidošanās vai lobīšanās (norāda uz Stīvensa-Džonsona sindromu)
- Acs krāsainās daļas iekaisums
Ziņošana par blakusparādībām
Ja Jums rodas jebkādas blakusparādības, konsultējieties ar ārstu vai farmaceitu. Tas attiecas arī uz iespējamām blakusparādībām, kas nav minētas šajā instrukcijā. Jūs varat ziņot par blakusparādībām arī tieši, izmantojot V pielikumā minēto valsts ziņošanas sistēmu. Ziņojot par blakusparādībām, jūs varat palīdzēt iegūt vairāk informācijas par šo zāļu drošumu.
Derīguma termiņš un saglabāšana
Uzglabāt šīs zāles bērniem neredzamā un nepieejamā vietā.
Nelietot šīs zāles pēc derīguma termiņa beigām, kas norādīts uz blistera un kastītes pēc “Derīgs līdz / EXP”. Derīguma termiņš attiecas uz mēneša pēdējo dienu.
Šīm zālēm nav nepieciešami īpaši uzglabāšanas apstākļi.
Neizmetiet zāles kanalizācijā vai sadzīves atkritumos. Jautājiet farmaceitam, kā izmest zāles, kuras vairs nelietojat. Tas palīdzēs aizsargāt vidi.
Sastāvs un zāļu forma
Ko Tarceva satur:
- Tarceva aktīvā viela ir erlotinibs. Katra apvalkotā tablete satur 25 mg, 100 mg vai 150 mg erlotiniba (erlotiniba hidrohlorīda veidā) atkarībā no stipruma.
- Citas sastāvdaļas ir: Tabletes kodols: laktozes monohidrāts, mikrokristāliskā celuloze, nātrija cietes glikolāts A tips, nātrija laurilsulfāts, magnija stearāts (laktozes monohidrātu skatīt arī 2. sadaļā). Tabletes apvalks: hipromeloze, hidroksipropilceluloze, titāna dioksīds, makrogols.
Tarceva ārējais izskats un iepakojums:
Tarceva 25 mg tiek piegādāta balta vai dzeltenīga, apaļa apvalkotā tablete, kuras vienā pusē ir iegravēts "T 25", un tā ir pieejama iepakojumā pa 30 tabletēm.
Tarceva 100 mg tiek piegādāta balta vai dzeltenīga, apaļa apvalkotā tablete, kuras vienā pusē ir iegravēts "T 100", un tā ir pieejama iepakojumā pa 30 tabletēm.
Tarceva 150 mg tiek piegādāta balta vai dzeltenīga, apaļa apvalkotā tablete, kuras vienā pusē ir iegravēts "T 150", un tā ir pieejama iepakojumā pa 30 tabletēm.
Avota lietošanas instrukcija: AIFA (Itālijas zāļu aģentūra). Saturs publicēts 2016. gada janvārī. Pašlaik pieejamā informācija var nebūt atjaunināta.
Lai piekļūtu visjaunākajai versijai, ieteicams piekļūt AIFA (Itālijas zāļu aģentūra) vietnei. Atruna un noderīga informācija.
01.0 ZĀĻU NOSAUKUMS
TARCEVA 150 MG TABLETES, KAS PĀRKLĀTAS AR Plēvi
02.0 KVALITATĪVAIS UN KVANTITATĪVAIS SASTĀVS
Viena apvalkotā tablete satur 150 mg erlotiniba (erlotiniba hidrohlorīda veidā).
Palīgvielas ar zināmu iedarbību: katra apvalkotā tablete satur 103,82 mg laktozes monohidrāta.
Pilnu palīgvielu sarakstu skatīt apakšpunktā 6.1.
03.0 ZĀĻU FORMA
Apvalkotā tablete.
Baltas līdz dzeltenīgas, apaļas, abpusēji izliektas tabletes ar iegravētu "T 150" vienā pusē.
04.0 KLĪNISKĀ INFORMĀCIJA
04.1 Terapeitiskās indikācijas
Nesīkšūnu plaušu karcinoma ( Nesīkšūnu plaušu vēzis (NSCLC):
Tarceva ir indicēts pirmās līnijas ārstēšanai pacientiem ar lokāli progresējošu vai metastātisku nesīkšūnu plaušu vēzi (NSCLC) ar aktivējošām EGFR mutācijām.
Tarceva ir indicēts arī kā pārejas terapija pacientiem ar lokāli progresējošu vai metastātisku NSCLC ar aktivējošām EGFR mutācijām un stabilu slimību pēc pirmās līnijas ķīmijterapijas.
Tarceva ir indicēts arī pacientu ar lokāli progresējošu vai metastātisku NSCLC ārstēšanai pēc vismaz viena iepriekšēja ķīmijterapijas režīma neveiksmes.
Izrakstot Tarceva, jāņem vērā faktori, kas saistīti ar dzīvildzes palielināšanos.
Ārstēšana neuzrādīja izdzīvošanas ieguvumus vai citus klīniski nozīmīgus efektus pacientiem ar epidermas augšanas faktora receptoru (EGFR) -IHC negatīviem audzējiem (skatīt 5.1. Apakšpunktu).
Aizkuņģa dziedzera vēzis:
Tarceva kombinācijā ar gemcitabīnu ir indicēts metastātiska aizkuņģa dziedzera vēža ārstēšanai.
Izrakstot Tarceva, jāņem vērā faktori, kas saistīti ar dzīvildzes palielināšanos (skatīt 4.2. Un 5.1. Apakšpunktu).
Nav pierādīta izdzīvošanas priekšrocība pacientiem ar lokāli progresējošu slimību.
04.2 Devas un lietošanas veids
Ārstēšana ar Tarceva jāuzrauga ārstam, kuram ir pieredze pretvēža terapijas lietošanā.
Pacienti ar nesīkšūnu plaušu karcinomu:
Pirms Tarceva terapijas uzsākšanas pacientiem ar ķīmijterapiju iepriekš neārstētiem pacientiem ar progresējošu vai metastātisku NSCLC jāveic EGFR mutācijas pārbaude.
Ieteicamā Tarceva dienas deva ir 150 mg, ko lieto vismaz vienu stundu pirms vai divas stundas pēc ēšanas.
Pacienti ar aizkuņģa dziedzera vēzi:
Ieteicamā Tarceva dienas deva ir 100 mg, kas jālieto vismaz vienu stundu pirms vai divas stundas pēc ēšanas kombinācijā ar gemcitabīnu (sk. Gemcitabīna zāļu aprakstu aizkuņģa dziedzera vēža indikācijā).
Pacientiem, kuriem pirmajās 4–8 terapijas nedēļās neveidojas izsitumi uz ādas, jāpārvērtē, vai turpināt ārstēšanu ar Tarceva (skatīt 5.1. Apakšpunktu).
Ja deva ir jāpielāgo, tā katru reizi jāsamazina par 50 mg (skatīt apakšpunktu 4.4).
Tarceva ir pieejams 25 mg, 100 mg un 150 mg stiprumos.
Vienlaicīgai CYP3A4 substrātu un modulatoru lietošanai var būt nepieciešams mainīt devas (skatīt 4.5. Apakšpunktu).
Pacienti ar aknu mazspēju: Erlotiniba eliminācija notiek, metabolizējoties aknās un izdaloties ar žulti. Lai gan erlotiniba iedarbība bija līdzīga pacientiem ar vidēji smagiem aknu darbības traucējumiem (Child-Pugh rādītājs 7-9) un pacientiem ar adekvātu aknu darbību, jāievēro piesardzība, lietojot Tarceva pacientiem ar aknu darbības traucējumiem. Ja rodas nopietnas blakusparādības, deva jāapsver Tarceva terapijas samazināšana vai pārtraukšana. Erlotiniba drošums un efektivitāte nav pētīta pacientiem ar smagiem aknu darbības traucējumiem (ASAT / SGOT un ALAT / SGPT> 5 x NAR). Tarceva nav ieteicams lietot pacientiem ar smagiem aknu darbības traucējumiem (skatīt 5.2. Apakšpunktu).
Pacienti ar nieru mazspēju: Erlotiniba drošība un efektivitāte nav pētīta pacientiem ar nieru mazspēju (kreatinīna līmenis serumā> 1,5 reizes pārsniedz normas augšējo robežu). Pamatojoties uz farmakokinētikas datiem, pacientiem ar vieglu vai vidēji smagu nieru mazspēju devas nav jāmaina ( skatīt 5.2. apakšpunktu). Tarceva lietošana pacientiem ar smagu nieru mazspēju nav ieteicama.
Pediatriskā populācija: Erlotiniba drošība un efektivitāte pacientiem līdz 18 gadu vecumam nav pierādīta.Tarceva lietošana bērniem nav ieteicama.
Smēķētāji: Ir pierādīts, ka cigarešu smēķēšana samazina erlotiniba iedarbību par 50–60%. Maksimālā pieļaujamā Tarceva deva pacientiem, kuri smēķē cigaretes ar NSCLC, bija 300 mg. Pacientiem nav noteikta ilgtermiņa efektivitāte un drošība, kas ir augstāka par sākotnēji ieteicamo devu. kuri turpina smēķēt (skatīt 4.5. un 5.2. apakšpunktu). Tāpēc smēķētājiem jāiesaka pārtraukt smēķēšanu, jo erlotiniba koncentrācija plazmā smēķētājiem ir zemāka nekā nesmēķētājiem.
04.3 Kontrindikācijas
Paaugstināta jutība pret erlotinibu vai kādu no 6.1. Apakšpunktā uzskaitītajām palīgvielām.
04.4 Īpaši brīdinājumi un piesardzība lietošanā
EGFR mutācijas statusa novērtējums:
Lai novērtētu pacienta EGFR mutācijas stāvokli, ir svarīgi izvēlēties labi apstiprinātu un stabilu metodiku, lai izvairītos no kļūdaini negatīviem vai kļūdaini pozitīviem konstatējumiem.
Smēķētāji
Smēķētājiem jāiesaka pārtraukt smēķēšanu, jo erlotiniba koncentrācija plazmā smēķētājiem ir zemāka nekā nesmēķētājiem. Samazinājuma pakāpe varētu būt klīniski nozīmīga (skatīt 4.5. Apakšpunktu).
Plaušu intersticiāla slimība (ILD))
Retāk pacientiem, kuri lietoja Tarceva nesīkšūnu plaušu vēža (NSCLC), aizkuņģa dziedzera vēža vai citu progresējošu cietu audzēju ārstēšanai, retos gadījumos ir ziņots par ILD līdzīgiem gadījumiem, dažkārt letāliem. NSCLC galvenajā pētījumā BR.21 ILD sastopamība (0,8%) bija vienāda placebo grupā un Tarceva grupā. Aizkuņģa dziedzera vēža pētījumā saistībā ar gemcitabīnu ILD līdzīgu notikumu biežums bija 2,5 % Tarceva un gemcitabīna grupā, salīdzinot ar 0,4% placebo un gemcitabīna grupā. Kopējais sastopamības biežums ar Tarceva ārstētiem pacientiem visos pētījumos (ieskaitot nekontrolētu un vienlaicīgu ķīmijterapiju) ir aptuveni 0,6%, salīdzinot ar 0,2% ar placebo ārstētiem pacientiem. Starp ziņotajām diagnozēm pacientiem, kuriem ir aizdomas par ILD līdzīgiem notikumiem pneimonija, staru pneimonija, paaugstinātas jutības pneimonīts, intersticiāla pneimonija, plaušu intersticiāla slimība, obliterārs bronhiolīts, plaušu fibroze, akūts elpošanas distresa sindroms, alveolīts un "plaušu infiltrācija. Simptomi parādījās dažu dienu vai vairāku mēnešu laikā pēc Tarceva terapijas uzsākšanas. Mulsinoši vai vienlaicīgi faktori, piemēram, vienlaicīga vai iepriekš veikta ķīmijterapija, iepriekšēja staru terapija, jau esoša parenhīmas plaušu slimība, metastāzes vai plaušu infekcijas. Biežāk sastopama ILD 1,5%) tika novērots japāņu izcelsmes iedzīvotāju vidū.
Pacientiem ar jauniem un / vai progresējošiem neizskaidrojamiem plaušu simptomiem, piemēram, aizdusu, klepu un drudzi, Tarceva lietošana jāpārtrauc, kamēr nav veikta diagnostiskā pārbaude. Pacienti, kuri vienlaikus tiek ārstēti ar erlotinibu un gemcitabīnu, rūpīgi jānovēro, vai nav iespējama ILD līdzīga toksicitāte. Ja tiek diagnosticēts ILD, Tarceva lietošana jāpārtrauc un pēc vajadzības jāsāk atbilstoša ārstēšana (skatīt 4.8. Apakšpunktu).
Caureja, dehidratācija, elektrolītu līdzsvara traucējumi un nieru mazspēja
Apmēram 50% pacientu, kuri tika ārstēti ar Tarceva, bija caureja (ieskaitot ļoti retus gadījumus ar letālu iznākumu); mērena vai smaga caureja jāārstē, piemēram, ar loperamīdu. Dažos gadījumos var būt nepieciešams samazināt devu. Klīniskajos pētījumos devas tika samazinātas par 50 mg. Devas samazināšana par 25 mg vienlaikus nav pētīta. Smagas vai ilgstošas caurejas, sliktas dūšas, anoreksijas vai vemšanas gadījumā, kas saistīta ar dehidratāciju, Tarceva lietošana jāpārtrauc un jāveic atbilstoši pasākumi dehidratācijas ārstēšanai (skatīt 4.8. Apakšpunktu). Ir ziņots par retiem hipokaliēmijas un nieru mazspējas gadījumiem (ieskaitot letālus gadījumus). Daži gadījumi bija sekundāri smagas dehidratācijas dēļ, ko izraisīja caureja, vemšana un / vai anoreksija, bet citi bija saistīti ar vienlaicīgu ķīmijterapiju.Smagākas vai ilgstošas caurejas vai dehidratācijas gadījumos, īpaši pacientu grupās ar pastiprinošiem riska faktoriem (īpaši saistībā ar ķīmijterapiju un citām zālēm, simptomiem vai slimībām vai citiem predisponējošiem apstākļiem, ieskaitot vecumu), Tarceva jāievada jāpārtrauc un jāveic atbilstoši pasākumi intensīvai pacienta intravenozai rehidratācijai. Turklāt pacientiem, kuriem ir dehidratācijas risks, jākontrolē nieru darbība un elektrolītu līmenis serumā, ieskaitot kāliju.
Hepatīts, aknu mazspēja
Ārstēšanas laikā ar Tarceva ziņots par retiem aknu mazspējas gadījumiem (ieskaitot letālus gadījumus). Iepriekš esoša aknu slimība vai hepatotoksisku zāļu vienlaicīga lietošana tika uzskatītas par traucējošiem faktoriem. Tādēļ šādiem pacientiem jāapsver periodiska aknu darbības pārbaude. Tarceva lietošana jāpārtrauc, ja aknu darbības traucējumi ir smagi (skatīt apakšpunktu 4.8). Pacientiem ar smagiem aknu darbības traucējumiem Tarceva lietošana nav ieteicama.
Kuņģa -zarnu trakta perforācija
Pacientiem, kuri lieto Tarceva, ir paaugstināts kuņģa un zarnu trakta perforācijas risks, kas novērots retāk (ieskaitot dažus letālus gadījumus). Risks ir lielāks pacientiem, kuri vienlaikus lieto antiangiogēnus līdzekļus, kortikosteroīdus, NPL un / vai uz taksānu balstītu ķīmijterapiju, vai kuriem ir bijusi peptiska čūla vai divertikulāra slimība. Pacientiem, kuriem attīstās kuņģa -zarnu trakta perforācija, ārstēšana ar Tarceva ir pilnībā jāpārtrauc (skatīt apakšpunktu 4.8).
Bullozi, eksfoliatīvi ādas bojājumi
Ir ziņots par bullozām, vezikulārām un eksfoliatīvām ādas slimībām, tostarp ļoti retos gadījumos, kas liecina par Stīvensa-Džonsona sindromu / toksisku epidermas nekrolīzi, kas dažos gadījumos bija letāla (skatīt 4.8. Apakšpunktu). Ārstēšana ar Tarceva jāpārtrauc vai jāpārtrauc, ja pacientam attīstās smagi bullozi, vezikulāri vai eksfoliatīvi traucējumi. Pacienti ar bullozām un eksfoliatīvām ādas izmaiņām jānovērtē attiecībā uz ādas infekcijām un jāārstē saskaņā ar vietējām vadlīnijām.
Acu patoloģijas
Pacientiem, kuriem ir pazīmes vai simptomi, kas liecina par keratītu, piemēram, akūts acu iekaisums vai acs pasliktināšanās, asarošana, fotofobija, neskaidra redze, acu sāpes un / vai sarkanas acis, nekavējoties jānosūta pie oftalmoloģijas speciālista. Ja tiek apstiprināta čūlaina keratīta diagnoze, ārstēšana ar Tarceva jāpārtrauc vai jāpārtrauc. Ja tiek diagnosticēts keratīts, rūpīgi jāapsver ārstēšanas turpināšanas ieguvumi un riski. Tarceva jālieto piesardzīgi pacientiem, kuriem anamnēzē ir keratīts, čūlainais keratīts vai smaga sausa acs. Kontaktlēcu lietošana ir arī keratīta un čūlas riska faktors. Lietojot Tarceva, ziņots par ļoti retiem radzenes perforācijas vai čūlas gadījumiem (skatīt apakšpunktu 4.8).
Mijiedarbība ar citām zālēm
Spēcīgi CYP3A4 induktori var samazināt erlotiniba efektivitāti, savukārt spēcīgi CYP3A4 inhibitori var palielināt toksicitāti. Jāizvairās no vienlaicīgas ārstēšanas ar šāda veida vielām (skatīt 4.5. Apakšpunktu).
Citi mijiedarbības veidi
Erlotinibu raksturo šķīdības samazināšanās pie pH vērtībām virs 5. Zāles, kas maina kuņģa -zarnu trakta augšdaļas (GI) pH, piemēram, protonu sūkņa inhibitori, H2 antagonisti un antacīdi, var ietekmēt erlotiniba šķīdību, un tāpēc tā bioloģiskā pieejamība. Tarceva devas palielināšana, ja to lieto vienlaikus ar šīm zālēm, var nekompensēt iedarbības samazināšanos. Jāizvairās no erlotiniba un protonu sūkņa inhibitoru kombinācijas. Erlotiniba un H2 antagonistu un antacīdu vienlaicīgas lietošanas ietekme nav zināma, tomēr ir iespējams, ka biopieejamība ir samazināta. Tādēļ jāizvairās no šo zāļu vienlaicīgas lietošanas. 4.5. apakšpunkts). Ja Tarceva terapijas laikā uzskata, ka antacīdi ir nepieciešami, tie jālieto vismaz 4 stundas pirms vai 2 stundas pēc Tarceva dienas devas.
Tabletes satur laktozi, un tās nedrīkst ievadīt pacientiem ar retu iedzimtu galaktozes nepanesību, Lapp laktāzes deficītu vai glikozes-galaktozes malabsorbciju.
04.5 Mijiedarbība ar citām zālēm un citi mijiedarbības veidi
Mijiedarbības pētījumi veikti tikai pieaugušajiem.
Erlotinibs un citi CYP substrāti
Erlotinibs ir spēcīgs CYP1A1 inhibitors un mērens CYP3A4 un CYP2C8 inhibitors, kā arī spēcīgs glikuronizācijas inhibitors in vitro ar UGT1A1.
Tā kā CYP1A1 ekspresija cilvēka audos ir ļoti samazināta, spēcīgās CYP1A1 inhibīcijas fizioloģiskā nozīme nav zināma.
Ja erlotinibu lietoja vienlaikus ar mērenu CYP1A2 inhibitoru ciprofloksacīnu, erlotiniba iedarbība [AUC] tika ievērojami palielināta par 39%, bet netika novērotas statistiski nozīmīgas Cmax izmaiņas. Aktīvā metabolīta iedarbība palielinājās par aptuveni 60% un 48% Attiecīgi AUC un C var samazināties erlotiniba daudzums.
Iepriekšēja ārstēšana vai Tarceva vienlaicīga lietošana nemainīja CYP3A4 prototipisko substrātu, piemēram, midazolāma un eritromicīna, klīrensu, bet, šķiet, samazināja midazolāma perorālo biopieejamību līdz 24%. Citā klīniskajā pētījumā erlotinibs nemainīja vienlaikus lietota paklitaksela CYP3A4 / 2C8 substrāta farmakokinētiku. Tāpēc maz ticama būtiska mijiedarbība ar citu CYP3A4 substrātu klīrensu.
Glikuronidācijas inhibīcija var izraisīt mijiedarbību ar zālēm, kas ir UGT1A1 substrāti un kuras tiek izvadītas tikai pa šo ceļu. Pacientiem ar samazinātu UGT1A1 ekspresijas līmeni vai ar ģenētiskām glikuronizācijas izmaiņām (piemēram, Gilberta slimība) var būt paaugstināta bilirubīna koncentrācija serumā, un viņi jāārstē piesardzīgi.
Cilvēkiem erlotinibs aknās tiek metabolizēts ar aknu citohromiem, galvenokārt ar CYP3A4 un mazākā mērā ar CYP1A2. Ārpus aknu metabolisms, ko ietekmē CYP3A4 zarnās, CYP1A1 plaušās un CYP1B1 audzēja audos, arī potenciāli veicina erlotiniba metaboliskais klīrenss. Iespējama mijiedarbība ar aktīvajām sastāvdaļām, kuras metabolizē šie fermenti vai kas iedarbojas uz tiem kā inhibitoriem vai induktoriem.
Spēcīgi CYP3A4 aktivitātes inhibitori samazina erlotiniba metabolismu un palielina erlotiniba koncentrāciju plazmā. Klīniskajā pētījumā vienlaikus lietojot erlotinibu un spēcīgu CYP3A4 inhibitoru ketokonazolu (200 mg divas reizes dienā 5 dienas), palielinājās erlotiniba iedarbība (86% AUC un 69% Cmax). Tādēļ jāievēro piesardzība, lietojot erlotinibu kombinācijā ar spēcīgu CYP3A4 inhibitoru, piemēram, azola pretsēnīšu līdzekļiem (piemēram, ketokonazolu, itrakonazolu, vorikonazolu), proteāzes inhibitoriem, eritromicīnu vai klaritromicīnu.erlotiniba deva, īpaši toksicitātes gadījumā.
Spēcīgi CYP3A4 aktivitātes induktori palielina erlotiniba metabolismu un ievērojami samazina erlotiniba koncentrāciju plazmā. Klīniskajā pētījumā vienlaicīga erlotiniba un rifampicīna (600 mg dienā perorāli 7 dienas), spēcīga CYP3A4 induktora, lietošana izraisīja vidējais erlotiniba AUC samazinājums par 69%. Vienlaicīgi lietojot rifampicīnu un vienu 450 mg Tarceva devu, vidējā erlotiniba iedarbība (AUC) bija par 57,5% lielāka nekā sasniedzot pēc vienas 150 mg Tarceva devas lietošanas ārstēšanas trūkums ar rifampicīnu. Tāpēc jāizvairās no Tarceva vienlaicīgas lietošanas ar CYP3A4 induktoriem. Pacientiem, kuriem nepieciešama vienlaicīga ārstēšana ar Tarceva un spēcīgu CYP3A4 induktoru, piemēram, rifampicīnu, jāapsver devas palielināšana līdz 300 mg, bet viņu drošība (ieskaitot nieru un aknu darbību un seruma elektrolītus) tiek rūpīgi uzraudzīta un, ja tā ir labi panesama ilgāk pēc 2 nedēļām, var apsvērt turpmāku devas palielināšanu līdz 450 mg, rūpīgi kontrolējot drošību. Iedarbības samazināšanās var notikt arī ar citiem induktoriem, piemēram, fenitoīnu, karbamazepīnu, barbiturātiem vai asinszāli (Hypericum perforatum). Lietojot šīs aktīvās vielas kopā ar erlotinibu, jāievēro piesardzība. Kad vien iespējams, jāapsver alternatīva ārstēšana bez spēcīgas induktīvas CYP3A4 aktivitātes.
Erlotinibs un no kumarīna iegūti antikoagulanti
Pacientiem, kuri lieto Tarceva, ziņots par mijiedarbības gadījumiem ar antikoagulantiem, kas iegūti no kumarīna, ieskaitot varfarīnu, kā rezultātā palielinājās INR (starptautiskā normalizētā attiecība) un asiņošanas gadījumi, kas dažos gadījumos bija letāli. jebkādas protrombīna laika vai INR izmaiņas.
Erlotinibs un statīni
Tarceva un statīna kombinācija var palielināt statīnu izraisītas miopātijas, tai skaitā rabdomiolīzes, risku, kas novērots reti.
Erlotinibs un smēķētāji
Farmakokinētiskās mijiedarbības pētījuma rezultāti parādīja ievērojamu samazinājumu par 2,8 pēc Tarceva ievadīšanas; Smēķētājiem, salīdzinot ar nesmēķētājiem, attiecīgi 1,5 un 9 reizes pārsniedz AUCinf, Cmax un 24 stundu plazmas koncentrāciju (skatīt 5.2. Apakšpunktu). Tādēļ pacienti, kuri joprojām smēķē, jāmudina pēc iespējas ātrāk pārtraukt smēķēšanu. uzsākt Tarceva terapiju, pretējā gadījumā erlotiniba koncentrācija plazmā samazinās. Samazinātas iedarbības klīniskā ietekme nav galīgi noteikta, taču tā var būt klīniski nozīmīga.
Erlotiniba un P-glikoproteīna inhibitori
Erlotinibs ir aktīvās sastāvdaļas nesēja P-glikoproteīna substrāts. Vienlaicīga P-glikoproteīnu inhibitoru, piemēram, ciklosporīna un verapamila, lietošana var izraisīt erlotiniba sadalījumu un / vai elimināciju. Šīs mijiedarbības sekas, piemēram, attiecībā uz CNS toksicitāti, nav noteiktas. Šādās situācijās vajadzētu rīkoties piesardzīgi.
Erlotinibs un zāles, kas maina pH
Erlotinibu raksturo šķīdības samazināšanās pie pH vērtībām virs 5. Zāles, kas maina kuņģa -zarnu trakta augšdaļas (GI) pH, var mainīt erlotiniba šķīdību un līdz ar to arī bioloģisko pieejamību. Erlotiniba vienlaicīga lietošana ar omeprazolu, protonu sūkņa inhibitoru (PPI), samazināja erlotiniba iedarbību [AUC] un maksimālo koncentrāciju [Cmax] attiecīgi par 46% un 61%. Tmax vai pussabrukšanas perioda izmaiņas netika konstatētas. Vienlaicīga Tarceva lietošana ar 300 mg ranitidīna, kas ir H2 receptoru antagonists, samazināja erlotiniba iedarbību [AUC] un maksimālo koncentrāciju [Cmax] attiecīgi par 33% un 54%. Palieliniet Tarceva devu, ja to lieto vienlaikus ar šīm zālēm, tas var nekompensēt iedarbības samazināšanai. Tomēr, ja Tarceva tika ievadīts pakāpeniski, 2 stundas pirms vai 10 stundas pēc 150 mg ranitidīna divreiz dienā, erlotiniba iedarbība [AUC] un maksimālā koncentrācija [Cmax] samazinājās tikai par 15% un 17% . Antacīdu ietekme uz erlotiniba uzsūkšanos nav pētīta, bet absorbcija var būt traucēta, kā rezultātā samazinās plazmas līmenis.Kopumā jāizvairās no erlotiniba kombinācijas ar protonu sūkņa inhibitoriem. Ja Tarceva terapijas laikā antacīdi tiek uzskatīti par nepieciešamiem, tie jālieto vismaz 4 stundas pirms vai 2 stundas pēc Tarceva dienas devas. Ja tiek apsvērts ranitidīns, abas zāles jāievada pakāpeniski: Tarceva jālieto plkst. vismaz 2 stundas pirms vai 10 stundas pēc ranitidīna ievadīšanas.
Erlotinibs un Gemcitabīns
Ib fāzes pētījumā netika novērota būtiska gemcitabīna ietekme uz erlotiniba farmakokinētiku, kā arī būtiska erlotiniba ietekme uz gemcitabīna farmakokinētiku.
Erlotinibs un karboplatīns / paklitaksela
Erlotinibs palielina platīna koncentrāciju. Klīniskajā pētījumā, lietojot erlotinibu vienlaikus ar karboplatīnu un paklitakselu, kopējā platīna AUC0-48 palielinājās par 10,6%. Lai gan šis pieaugums ir statistiski nozīmīgs, šīs atšķirības lielums netiek uzskatīts par klīniski nozīmīgu. Klīniskajā praksē var būt arī citi līdzfaktori, kas palielina karboplatīna iedarbību, piemēram, nieru mazspēja. Karboplatīna vai paklitaksela ietekmi uz erlotiniba farmakokinētiku.
Erlotinibs un kapecitabīns
Kapecitabīns var palielināt erlotiniba koncentrāciju.Lietojot erlotinibu vienlaikus ar kapecitabīnu, statistiski nozīmīgs erlotiniba AUC pieaugums un neliels Cmax pieaugums, salīdzinot ar vērtībām, kas novērotas citā pētījumā, kurā tika lietots tikai erlotinibs. Nav būtiskas erlotiniba ietekmes uz farmakokinētiku kapecitabīna.
Erlotinibs un proteasomu inhibitori
Saistībā ar darbības mehānismu proteasomu inhibitori, ieskaitot bortezomibu, var ietekmēt EGFR inhibitoru, tostarp erlotiniba, aktivitāti. Šo ietekmi apstiprina ierobežota klīnisko un preklīnisko datu pieejamība, kas izceļ EGFR noārdīšanos caur proteasomu.
04.6 Grūtniecība un zīdīšana
Grūtniecība
Nav pietiekamu datu par erlotiniba lietošanu grūtniecēm. Pētījumi ar dzīvniekiem neuzrādīja teratogenitāti vai patoloģiskas dzemdības. Tomēr nevar izslēgt negatīvu ietekmi uz grūtniecību, jo pētījumi ar žurkām un trušiem ir parādījuši palielinātu embriju / augļa mirstību (sk. apakšpunkts) Potenciālais risks cilvēkiem nav zināms.
Sievietes reproduktīvā vecumā
Sievietēm reproduktīvā vecumā jāiesaka izvairīties no grūtniecības ārstēšanas laikā ar Tarceva. Ārstēšanas laikā un vismaz 2 nedēļas pēc ārstēšanas beigām jāizmanto atbilstošas kontracepcijas metodes. Grūtniecēm ārstēšana jāturpina tikai gadījumos, kad iespējamais ieguvums mātei atsver risku auglim.
Barošanas laiks
Nav zināms, vai erlotinibs izdalās mātes pienā. Ņemot vērā iespējamo kaitējumu jaundzimušajam, mātēm jāiesaka barot bērnu ar krūti Tarceva terapijas laikā.
Auglība
Pētījumi ar dzīvniekiem neuzrādīja auglības traucējumus. Tomēr nevar izslēgt negatīvu ietekmi uz auglību, jo pētījumi ar dzīvniekiem liecina par ietekmi uz reproduktīvajiem parametriem (skatīt 5.3. Apakšpunktu). Iespējamais risks cilvēkiem nav zināms.
04.7 Ietekme uz spēju vadīt transportlīdzekļus un apkalpot mehānismus
Nav veikti pētījumi par spēju vadīt transportlīdzekļus un apkalpot mehānismus; tomēr erlotinibs nav saistīts ar garīgo spēju traucējumiem.
04.8 Nevēlamās blakusparādības
Nesīkšūnu plaušu vēzis (Tarceva lieto atsevišķi):
Randomizētā dubultmaskētā pētījumā (BR.21; Tarceva, kas tika lietots kā otrās līnijas terapija), visbiežāk ziņotās blakusparādības bija izsitumi (75%) un caureja (54%), vairumā gadījumu intensitāte bija vienāda ar 1. pakāpi /2 un pārvaldāms bez jebkādas iejaukšanās. 3/4 pakāpes izsitumi un caureja radās attiecīgi 9% un 6% pacientu, kuri tika ārstēti ar Tarceva, un abos gadījumos pētījums tika pārtraukts 1% pacientu, devu attiecīgi 6% un 1% pacientu. Pētījumā BR.21 vidējais laiks līdz izsitumiem bija 8 dienas un vidējais laiks līdz caurejas sākumam bija 12 dienas.
Kopumā izsitumi izpaužas kā "viegls vai vidēji smags eritematozs un papulārs-pustulārs izvirdums, kas var rasties vai pasliktināties vietās, kas pakļautas saules iedarbībai. Pacientiem, kuri pakļauti saules iedarbībai, var būt ieteicams lietot aizsargapģērbu" un / vai sauļošanās līdzekļi (piemēram, pamatojoties uz minerālvielām).
1. tabulā ir apkopotas pēc NCI-CTC pakāpes (Nacionālā vēža institūta kopējie toksicitātes kritēriji) nevēlamās blakusparādības, kas ar Tarceva ārstētiem pacientiem, biežāk sastopamas (≥3%), salīdzinājumā ar placebo grupu, tika veiktas pamatpētījumā BR.21 un vismaz 10% pacientu Tarceva grupā.
Lai klasificētu nevēlamās blakusparādības pēc biežuma, tiek izmantoti šādi termini: ļoti bieži (≥1 / 10); bieži (≥ 1/100,
Katrā sastopamības biežuma grupā blakusparādības ir norādītas to smaguma pakāpes samazināšanās secībā.
1. tabula. Ļoti bieži novērotās blakusparādības pētījumā BR.21
* Smagas infekcijas ar vai bez neitropēnijas, ieskaitot pneimoniju, sepsi un celulītu.
** Var izraisīt dehidratāciju, hipokaliēmiju un nieru mazspēju.
*** Izsitumi ietvēra aknveida dermatīta gadījumus.
2 citos randomizētos, dubultmaskētos, placebo kontrolētos III fāzes pētījumos BO18192, (SATURN) un BO25460 (IUNO); Tarceva tika ievadīta uzturošā terapijā pēc pirmās līnijas ķīmijterapijas. Šie pētījumi tika veikti kopā 1532 pacientiem ar progresējošu, recidivējošu vai metastātisku NSCLC pēc standarta pirmās līnijas ķīmijterapijas, kas balstīta uz platīnu, netika atklāti jauni drošības ziņojumi.
Visbiežāk novērotās blakusparādības ar Tarceva ārstētiem pacientiem pētījumos BO18192 un BO25460 bija izsitumi un caureja (skatīt 2. tabulu). Nevienā pētījumā netika novēroti 4. pakāpes izsitumi vai caureja. Izsitumi un caureja izraisīja Tarceva lietošanas pārtraukšanu attiecīgi 1% un 1% gadījumu
2. tabula. Visbiežāk novērotās blakusparādības pētījumos BO18192 (SATURN) un BO25460 (IUNO)
* Drošības novērtēšanas grupa
Atklātā III fāzes pētījumā ML 20650 154 pacientiem Tarceva drošība pirmās līnijas ārstēšanai pacientiem ar NSCLC un EGFR aktivējošām mutācijām tika novērtēta 75 pacientiem; netika atklāti jauni saistīti ziņojumi.
Visbiežāk novērotās nevēlamās blakusparādības ar Tarceva ārstētiem pacientiem ar ML 20650 bija izsitumi un caureja (jebkura pakāpe, attiecīgi 80% un 57%), pārsvarā 1. un 2. pakāpes intensitāte, kas bija ārstējama. 3. pakāpes izsitumi un caureja radās 9% un 4 attiecīgi % pacientu. Netika novēroti 4. pakāpes izsitumi vai caureja. Izsitumi un caureja izraisīja Tarceva lietošanas pārtraukšanu 1 % pacientu. Devas pielāgošana (pārtraukšana vai samazināšana) izsitumu un caurejas dēļ bija nepieciešama attiecīgi 11% un 7% pacientu.
Aizkuņģa dziedzera vēzis (Tarceva lieto vienlaikus ar gemcitabīnu):
Visizplatītākās nevēlamās blakusparādības galvenajā pētījumā PA.3 aizkuņģa dziedzera vēža slimniekiem, kuri tika ārstēti ar Tarceva 100 mg un gemcitabīnu, bija nogurums, izsitumi un caureja. Tarceva plus gemcitabīna grupā izsitumi un 3/4 pakāpes caureja tika ziņoti 5% pacientu. Vidējais laiks līdz izsitumiem un caurejai bija attiecīgi 10 dienas un 15 dienas. Izsitumi un caureja izraisīja devu samazināšanu 2% pacientu, un pētījums tika pārtraukts līdz 1% pacientu, kuri saņēma Tarceva un gemcitabīnu.
Galvenajā pētījumā PA.3 blakusparādības, kas parādījās biežāk (≥3%) pacientiem, kuri tika ārstēti ar Tarceva 100 mg kopā ar gemcitabīnu, nekā placebo un gemcitabīna grupā, un vismaz 10% pacientu Tarceva grupā 100 mg plus gemcitabīns ir apkopots 3. tabulā, pamatojoties uz Nacionālā vēža institūta kopējiem toksicitātes kritērijiem (NCI-CTC).
Lai klasificētu nevēlamās blakusparādības pēc biežuma, tiek izmantoti šādi termini: ļoti bieži (≥1 / 10); bieži (≥ 1/100,
Katrā sastopamības biežuma grupā blakusparādības ir norādītas to smaguma pakāpes samazināšanās secībā.
3. tabula. Ļoti biežas blakusparādības pētījumā PA.3 (100 mg kohorta)
* Smagas infekcijas ar vai bez neitropēnijas, ieskaitot pneimoniju, sepsi un celulītu.
** Var izraisīt dehidratāciju, hipokaliēmiju un nieru mazspēju.
*** Izsitumi ietvēra aknveida dermatīta gadījumus.
Citas piezīmes:
Tarceva drošuma novērtējums ir balstīts uz datiem no vairāk nekā 1500 pacientiem, kuri tika ārstēti ar vismaz vienu 150 mg Tarceva devu atsevišķi, un vairāk nekā 300 pacientiem, kuri tika ārstēti ar Tarceva 100 vai 150 mg kombinācijā ar gemcitabīnu.
Pacientiem, kuri tika ārstēti ar Tarceva monoterapiju, un pacientiem, kuri vienlaikus tika ārstēti ar ķīmijterapiju, tika novērotas šādas blakusparādības.
Ļoti bieži novērotās nevēlamās blakusparādības pētījumos BR 21 un PA 3 ir aprakstītas 1. un 3. tabulā, citas blakusparādības, ieskaitot blakusparādības, kas apkopotas citos pētījumos, ir apkopotas 4. tabulā.
Katrā sastopamības biežuma grupā blakusparādības ir norādītas to smaguma pakāpes samazināšanās secībā.
4. tabula. Nevēlamās blakusparādības pēc biežuma kategorijas
1 Pētījumā PA.3.
2 Tostarp skropstas, kas aug iekšā, pārmērīga skropstu augšana un sabiezēšana.
3 Ieskaitot letālus gadījumus, pacientiem, kuri Tarceva lieto NSCLC vai citu progresējošu cietu audzēju ārstēšanai (skatīt apakšpunktu 4.4). Biežums tika novērots japāņu izcelsmes pacientiem.
4 Klīniskajos pētījumos daži gadījumi ir saistīti ar vienlaicīgu varfarīna lietošanu (skatīt 4.5. Apakšpunktu) un dažreiz ar NPL vienlaicīgu lietošanu.
5 Ieskaitot alanīna aminotransferāzes (ALAT), aspartātaminotransferāzes (ASAT) un bilirubīna līmeņa paaugstināšanos). Tās visbiežāk bija vieglas vai vidēji smagas, pārejošas vai saistītas ar metastāzēm aknās.
6 Ieskaitot letālus gadījumus. Jau esoša aknu slimība vai vienlaicīga hepatotoksisku zāļu lietošana tika uzskatītas par traucējošiem faktoriem (skatīt apakšpunktu 4.4).
7 Tostarp letāli gadījumi (skatīt apakšpunktu 4.4).
Ziņošana par iespējamām blakusparādībām
Ir svarīgi ziņot par iespējamām blakusparādībām, kas radušās pēc zāļu reģistrācijas, jo tas ļauj nepārtraukti uzraudzīt zāļu ieguvuma un riska attiecību. Veselības aprūpes speciālistus lūdz ziņot par visām iespējamām blakusparādībām, izmantojot valsts ziņošanas sistēmu. "Adrese https: //www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
04.9 Pārdozēšana
Simptomi
Veseliem indivīdiem ir panesamas vienreizējas Tarceva devas līdz 1000 mg erlotiniba un vēža slimniekiem līdz 1600 mg. Atkārtotas 200 mg devas divas reizes dienā veseliem cilvēkiem pēc dažām lietošanas dienām bija slikti panesamas. Pamatojoties uz šo pētījumu datiem, iespējams, ka, lietojot lielākas devas nekā ieteicamas, var rasties nopietnas blakusparādības, piemēram, caureja, izsitumi un, iespējams, paaugstināta aknu aminotransferāzes aktivitāte.
Vadība
Ja ir aizdomas par pārdozēšanu, Tarceva lietošana jāpārtrauc un jāsāk simptomātiska ārstēšana.
05.0 FARMAKOLOĢISKĀS ĪPAŠĪBAS
05.1 Farmakodinamiskās īpašības
Farmakoterapeitiskā grupa: proteīna kināzes inhibitoru pretaudzēju zāles.
ATĶ kods: L01XE03.
Darbības mehānisms
Erlotinibs ir epidermas augšanas faktora receptors / I tipa cilvēka epidermas augšanas faktora receptors (EGFR, pazīstams arī kā HER1) tirozīnkināzes inhibitors. Erlotinibs ir spēcīgs EGFR intracelulārās fosforilācijas inhibitors. EGFR tiek izteikts uz normālu un audzēja šūnu virsmas. Neklīniskajos modeļos EGFR fosfotirozīna inhibīcija izraisa šūnu stāzi un / vai nāvi.
EGFR mutācijas var izraisīt "antiapoptotisku un proliferējošu signalizācijas ceļu konstitutīvu aktivizēšanu. Erlotiniba spēcīgā efektivitāte, bloķējot EGFR mediēto signālu pārraidi šajos EGFR mutācijas pozitīvajos audzējos, ir saistīta ar ciešu saikni starp erlotinibu ATP saistīšanās vietā". EGFR mutācijas kināzes domēnā.Pakārtotā transdukcijas signāla blokādes dēļ šūnu proliferācija tiek apturēta un šūnu nāve tiek izraisīta caur iekšējo apoptotisko ceļu. Audzēja regresija tiek novērota peles modeļos ar izteiktu šo aktivizējošo EGFR mutāciju ekspresiju.
Klīniskā efektivitāte
• Nesīkšūnu plaušu vēža (NSCLC) pirmās rindas terapija pacientiem ar aktivējošām EGFR mutācijām (Tarceva lieto atsevišķi):
Tarceva efektivitāte pirmās rindas pacientiem ar aktivējošām EGFR mutācijām NSCLC tika pierādīta nejaušinātā, atklātā III fāzes pētījumā (ML20650, EURTAC). Šis pētījums tika veikts ar kaukāziešu izcelsmes pacientiem ar metastātisku vai lokāli progresējošu NSCLC (IIIB un IV stadija), kuri iepriekš nebija saņēmuši ķīmijterapiju vai jebkādu sistēmisku pretvēža terapiju slimības dēļ un kuriem bija mutācijas EGFR tirozīnkināzes domēnā (eksona dzēšana). vai 21. eksona mutācija) Pacienti tika randomizēti 1: 1, lai saņemtu Tarceva 150 mg dienā vai līdz 4 cikliem ar platīna saturošu breketu ķīmijterapiju.
Primārais parametrs bija pētnieka novērtēts PFS.
Efektivitātes rezultāti ir apkopoti 5. tabulā.
5. tabula. Tarceva efektivitātes rezultāti pret ķīmijterapiju pētījumā ML20650 (EURTAC)
CR = pilnīga reakcija; RP = daļēja reakcija.
* Tika novērots slimības progresēšanas vai nāves riska samazinājums par 58%.
** Kopējais pētnieku un IRC novērtējums par PFS bija 70%.
*** "Augsts krustošanās ātrums" tika novērots 82% pacientu ķīmijterapijas grupā, kuri saņēma turpmāku terapiju ar EGFR saistītu tirozīnkināzes inhibitoru, un visi pacienti, izņemot 2, tika ārstēti ar Tarceva.
- NSCLC uzturošā terapija pēc pirmās līnijas ķīmijterapijas (Tarceva lieto monoterapijā):
Tarceva efektivitāte un drošība kā uzturošā terapija pēc pirmās līnijas ķīmijterapijas NSCLC tika pētīta randomizētā, dubultmaskētā, placebo kontrolētā pētījumā (BO18192, SATURN). Šajā pētījumā piedalījās 889 pacienti ar lokāli progresējošu NSCLC, kas pēc tam nebija progresējis. 4 divu līdzekļu, uz platīnu balstītas ķīmijterapijas cikli. Pacienti tika randomizēti 1: 1, lai ārstētu Tarceva 150 mg vai placebo iekšķīgi vienu reizi dienā līdz progresēšanai. L "Pētījuma galvenais mērķa kritērijs ietvēra dzīvildzi bez slimības progresēšanas (PFS) pacientiem. Demogrāfiskie un slimības raksturlielumi ievadīšanas brīdī bija labi līdzsvaroti starp abām ārstēšanas grupām.Pētījumā netika iekļauti pacienti ar ECOG PS> 1, nozīmīgas aknu vai nieru blakusslimības.
Šajā pētījumā visa populācija parādīja ieguvumu PFS primārajam mērķa kritērijam (HR = 0,71p)
67% ar placebo ārstēto pacientu EGFR mutācijas pozitīvā apakšgrupā saņēma EGFR-TKI inhibitorus otrajā vai turpmākajās ārstēšanas līnijās.
Pētījums BO25460 (IUNO) tika veikts 643 pacientiem ar progresējošu NSCLC bez EGFR aktivējošām mutācijām (19. eksona dzēšana vai 21. eksona L858R mutācija) un kuriem pēc četriem platīna ķīmijterapijas kursiem slimība nebija progresējusi.
Pētījuma mērķis bija salīdzināt erlotiniba kopējo dzīvildzi, kas tika ievadīta kā pirmā uzturēšanas līnija, un erlotinibu, kas tika ievadīts slimības progresēšanas laikā.Pētījums neatbilda primārajam mērķa kritērijam. Tarceva OS uzturēšanas laikā nebija pārāka par Tarceva, kas tika lietota kā otrās līnijas ārstēšana pacientiem, kuru audzējam nebija aktivizējošas EGFR mutācijas (HR = 1,02, 95% TI, 0,85-1, 22, p = 0,82). izdzīvošanas bez slimības progresēšanas (PFS) sekundārais mērķa kritērijs starp Tarceva un placebo uzturēšanas terapijas laikā neatšķīrās (HR = 0,94, 95% TI, 0,80-1,11; p = 0,48).
Pamatojoties uz pētījuma BO25460 (IUNO) datiem, Tarceva lietošana nav ieteicama kā pirmās izvēles uzturošā terapija pacientiem bez aktivizējošas EGFR mutācijas.
- NSCLC ārstēšana pēc vismaz vienas iepriekšējās ķīmijterapijas līnijas neveiksmes (Tarceva lieto atsevišķi):
Tarceva kā otrās / trešās līnijas terapijas efektivitāte un drošums tika pierādīts randomizētā, dubultmaskētā, placebo kontrolētā pētījumā (BR.21), kurā piedalījās 731 pacients ar lokāli progresējošu vai metastātisku NSŠP pēc vismaz ķīmijterapijas režīma neveiksmes. tika randomizēti 2: 1 pret ārstēšanu ar Tarceva 150 mg vai placebo perorāli vienu reizi dienā. Pētījuma mērķa kritēriji ietvēra kopējo dzīvildzi, izdzīvošanu bez slimības progresēšanas (PFS), atbildes reakciju, atbildes reakcijas ilgumu, laiku līdz ar plaušu vēzi saistīto simptomu (klepus, aizdusa) pasliktināšanos. un sāpes) un drošība. Primārais parametrs bija izdzīvošana.
Demogrāfiskās īpašības abās ārstēšanas grupās bija labi līdzsvarotas. Apmēram divas trešdaļas pacientu bija vīrieši, apmēram trešdaļai ECOG veiktspējas statusa (PS) bija 2 un 9% ECOG PS uzņemšanas laikā. 93% un 92% no visiem pacientiem. Tarceva grupa un placebo grupa iepriekš tika ārstēti ar shēmu, kuras pamatā bija platīns, un attiecīgi 36% un 37% no visiem pacientiem iepriekš bija ārstēti ar taksāniem.
Pielāgotā riska attiecība (HR) nāves gadījumiem Tarceva grupā, salīdzinot ar placebo grupu, bija 0,73 (95% TI: 0,60–0,87) (p = 0,001). 31,2% un 21,5% pacientu Tarceva un placebo grupās bija dzīvi pēc 12 mēnešiem. Vidējā kopējā dzīvildze Tarceva grupā bija 6,7 mēneši (95% TI: 5,5-7,8 mēneši) salīdzinājumā ar 4,7 mēnešiem placebo grupā (95% TI: 4,1-6, 3 mēneši).
Ietekme uz kopējo dzīvildzi tika pētīta dažādās pacientu apakšgrupās. Tarceva ietekme uz kopējo dzīvildzi bija līdzīga pacientiem ar sākotnējo darbības stāvokli (ECOG) 2-3 (HR = 0,77, TI 95 % 0,6-1,0) vai 0- 1 (HR = 0,73, 95% TI 0,6–0,9), vīriešiem (HR = 0,76, 95% TI 0, 6–0,9) vai sievietēm (HR = 0,80, 95% TI 0,6–1,1), pacientiem, kas jaunāki par 65 gadu vecumam (HR = 0,75, 95% TI 0,6–0,9) vai vecākiem pacientiem (HR = 0,79, 95% TI 0,6–1,0), pacientiem, kas ārstēti ar iepriekšēju shēmu (HR = 0,76, 95% TI% 0,6) -1,0) vai ar vairākām iepriekšējām shēmām (HR = 0,75, 95% TI 0,6-1,0), kaukāziešu izcelsmes pacientiem (HR = 0,79, 95% TI 0,6-1,0) vai aziātiem (HR = 0,61, 95% TI 0,4- 1,0), pacientiem ar adenokarcinomu (HR = 0,71, 95% TI 0,6-0, 9) vai plakanšūnu karcinomu (HR = 0,67, 95% TI 0,5-0,9), bet ne pacientiem ar citu histoloģiju (HR 1,04, 95 % TI 0,7-1,5), pacientiem ar IV stadijas slimību pie diagnozes (HR = 0,92, 95% TI 0,7-1,2) vai stadijā
45% pacientu ar zināmu EGFR ekspresijas stāvokli riska pakāpe bija 0,68 (95% TI 0,49-0,94) pacientiem ar EGFR pozitīviem audzējiem un 0, 93 (95% TI 0,63-1,36) pacientiem ar EGFR negatīviem audzējiem (definējis IHC, izmantojot EGFR pharmDx komplektu, kā EGFR negatīvs tiem, kuriem ir mazāk nekā 10% audzēja šūnu marķējuma). Atlikušajos 55% pacientu ar nezināmu EGFR ekspresijas stāvokli riska attiecība bija 0,77 (95% TI 0,61-0,98).
Vidējais PFS bija 9,7 nedēļas Tarceva grupā (95% TI, 8,4-12,4 nedēļas) salīdzinājumā ar 8,0 nedēļām placebo grupā (95% TI, 7,9-8,1 nedēļa).).
Tarceva grupā RECIST objektīvās atbildes reakcijas rādītājs bija 8,9% (95% TI, 6,4-12,0). Pirmie 330 pacienti tika novērtēti centralizēti (atbildes reakcijas koeficients 6, 2%); 401 pacients tika novērtēts kā pētnieks (atbildes reakcijas rādītājs 11,2%). .
Vidējais atbildes reakcijas ilgums bija 34,3 nedēļas, vismaz 9,7 un maksimāli 57,6+ nedēļas. 44,0% pacientu sasniedza pilnīgu, daļēju atbildes reakciju vai slimības stabilizāciju Tarceva grupā, salīdzinot ar 27,5% pacientu placebo grupā (p = 0,004).
Tarceva izdzīvošanas ieguvums tika novērots arī pacientiem, kuri nesasniedza objektīvu audzēja reakciju (RECIST kritēriji). Par to liecināja nāves riska koeficients 0,82 (95% TI, 0,68–0,99) pacientiem, kuri sasniedza slimības stabilizāciju vai progresēšanu kā labāko atbildes reakciju.
Salīdzinot ar placebo, Tarceva izraisīja simptomātiskus ieguvumus, ievērojami paildzinot laiku līdz klepus, aizdusa un sāpju pasliktināšanai.
- Aizkuņģa dziedzera vēzis (Tarceva vienlaikus ar gemcitabīnu ievadīts pētījumā PA.3):
Tarceva efektivitāte un drošība kombinācijā ar gemcitabīnu kā pirmās izvēles terapiju tika novērtēta randomizētā, dubultmaskētā, placebo kontrolētā pētījumā, kurā piedalījās pacienti ar lokāli progresējošu, neizoperējamu vai metastātisku aizkuņģa dziedzera vēzi. katru dienu nepārtraukti un iv gemcitabīns (1000 mg / m2, 1. cikls - 8. nedēļu cikla 1., 8., 15., 22., 29., 36. un 43. diena; 2. un nākamie cikli - 1., 8. un 15. diena) 4 nedēļu cikla laikā [apstiprināta aizkuņģa dziedzera vēža deva un shēma, skat. gemcitabīna zāļu aprakstu]]. Tarceva vai placebo lietoja iekšķīgi vienu reizi dienā līdz slimības progresēšanai vai nepieņemamai toksicitātei. Primārais parametrs bija kopējā dzīvildze.
Pacientu demogrāfiskie un slimības raksturlielumi ievadīšanas brīdī bija līdzīgi abām ārstēšanas grupām - Tarceva 100 mg plus gemcitabīns vai placebo plus gemcitabīns, izņemot nedaudz lielāku sieviešu daļu erlotiniba / gemcitabīna grupā, salīdzinot ar placebo / gemcitabīna grupu.
Izdzīvošana tika novērtēta plānotajā ārstēšanā, pamatojoties uz izdzīvošanas datiem. Rezultāti ir aprakstīti nākamajā tabulā (rezultāti pacientu grupām ar metastātisku un lokāli progresējošu slimību ir iegūti no izpētes apakšgrupu analīzes).
Pacienti, kuriem sākotnēji bija labvēlīgs klīniskais stāvoklis (zema sāpju intensitāte, labs QoL un labs PS), varētu gūt lielāku labumu no Tarceva, kā parādīts "post-hoc analīzē. Ieguvums galvenokārt izriet no zemas sāpju intensitātes līmeņa .
Post-hoc analīzē ar Tarceva ārstētiem pacientiem, kuriem attīstījās izsitumi, bija ilgāka kopējā dzīvildze, salīdzinot ar pacientiem, kuriem izsitumi neizraisījās (vidējā OS 7,2 mēneši pret 5 mēnešiem, HR: 0,61).
90% pacientu, kuri lietoja Tarceva, pirmajās 44 dienās parādījās izsitumi. Vidējais laiks līdz izsitumiem bija 10 dienas.
Pediatriskā populācija
Eiropas Zāļu aģentūra ir atbrīvojusi no pienākuma iesniegt pētījumu rezultātus ar Tarceva visās pediatriskās populācijas apakšgrupās indikācijām nesīkšūnu plaušu vēža un aizkuņģa dziedzera vēža gadījumā (informāciju par lietošanu bērniem skatīt 4.2. Apakšpunktā).
05.2 "Farmakokinētiskās īpašības
UzsūkšanāsMaksimālā erlotiniba koncentrācija plazmā tiek sasniegta aptuveni 4 stundas pēc iekšķīgas lietošanas. Pētījumā ar veseliem brīvprātīgajiem tika konstatēts, ka absolūtā biopieejamība ir 59%. Pārtika var palielināt iedarbību pēc iekšķīgas lietošanas.
Izplatīšana: erlotiniba vidējais šķietamais izkliedes tilpums ir 232 l, un tas izplatās cilvēka audzēja audos. Pētījumā, kas tika veikts ar 4 pacientiem (3 ar nesīkšūnu plaušu vēzi (NSCLC) un 1 ar balsenes vēzi), kuri tika ārstēti ar 150 mg Tarceva dienā perorāli, paraugi, kas iegūti, ķirurģiski izgriežot audzēju 9. dienā ārstēšanas laikā erlotiniba koncentrācija audzējā bija vidēji 1,185 ng / g audu, kas kopumā atbilst vidēji 63% (diapazons: 5-161%) no maksimālās koncentrācijas plazmā, kas novērota līdzsvara stāvoklī. Galvenās metabolītu aktīvās vielas bija sastopams audzējā koncentrācijā vidēji 160 ng / g audu, kas kopumā atbilst vidēji 113%(diapazons: 88-130%) no maksimālās koncentrācijas plazmā, kas novērota līdzsvara stāvoklī.Saistīšanās ar plazmas olbaltumvielām ir aptuveni 95%. Erlotinibs saistās ar seruma albumīnu un alfa-1 skābo glikoproteīnu (AAG).
BiotransformācijaCilvēkiem erlotinibu aknās metabolizē citohromi, galvenokārt CYP3A4 un mazākā mērā CYP1A2.Ekstrahepatiskais metabolisms, ko veicina CYP3A4 zarnās, CYP1A1 plaušās un 1B1 audzēja audos, potenciāli veicina erlotiniba metabolisko klīrensu.
Ir identificēti trīs galvenie vielmaiņas ceļi: 1) vienas vai abu sānu ķēžu O-demetilēšana, kam seko oksidēšana līdz karbonskābēm; 2) acetilēnās frakcijas oksidēšana, kam seko hidrolīze līdz arilkarbonskābei; un 3) fenilacetilēna aromātiskā hidroksilēšana. frakcija. Galvenie erlotiniba metabolīti OSI-420 un OSI-413, kas iegūti, veicot vienas no sānu ķēžu O-demetilēšanu, neklīniskajās analīzēs uzrādīja līdzīgu iedarbību kā erlotinibam. in vitro un audzēju modeļos in vivo. To koncentrācija plazmā ir mazāka par 10% no erlotiniba līmeņa, un tiem ir līdzīga erlotiniba farmakokinētika.
Eliminācija: erlotinibs galvenokārt tiek izvadīts ar fēcēm (> 90%), bet eliminācija caur nierēm veido tikai nelielu daļu (aptuveni 9%) no iekšķīgi ievadītā daudzuma. Mazāk nekā 2%no iekšķīgi lietotās devas izdalās nemainītā veidā. Viena populācijas farmakokinētiskā analīze no 591 pacientam, kuri tika ārstēti ar Tarceva monoterapiju, vidējais šķietamais klīrenss bija 4,47 l / h, un vidējais pussabrukšanas periods bija 36,2 stundas.
Farmakokinētika īpašās populācijās:
Pamatojoties uz populācijas farmakokinētikas analīzi, netika novērotas klīniski nozīmīgas korelācijas starp šķietamo paredzamo klīrensu un pacienta vecumu, ķermeņa svaru, dzimumu vai etnisko piederību. Ar pacientu saistīti faktori, kas parādīja korelāciju ar erlotiniba farmakokinētiku, bija kopējais bilirubīns, "AAG un" Palielināta kopējā bilirubīna un AAG koncentrācija serumā bija saistīta ar samazinātu erlotiniba klīrensu. Šo atšķirību klīniskā nozīme nav skaidra. Tomēr smēķētājiem palielinājās erlotiniba klīrensa ātrums.
To apstiprināja farmakokinētikas pētījums ar veseliem nesmēķētājiem un cigarešu smēķētājiem, kuri saņēma vienu 150 mg perorālu erlotinba devu. Cmax ģeometriskais vidējais bija 1056 ng / ml nesmēķētājiem un 689 ng / ml smēķētājiem ar vidējo smēķētāju un nesmēķētāju attiecību 65,2% (95% TI: 44,3 - 95,9; p = 0,031). AUC0 -inf ģeometriskais vidējais bija 18726 ng • h / ml nesmēķētājiem un 6718 ng • h / ml smēķētājiem ar vidējo attiecību 35,9% (95% TI: 23,7 - 54, 3; p
Pivotālajā III fāzes NSCLC pētījumā smēķētāji sasniedza erlotiniba līdzsvara koncentrāciju plazmā 0,65 μg / ml (n = 16), kas ir aptuveni 2 reizes zemāka nekā bijušajiem smēķētājiem vai pacientiem, kuri nekad nav smēķējuši (1,28 mcg / ml, n = 108). Šo efektu papildināja erlotiniba šķietamā plazmas klīrensa palielināšanās par 24%. I fāzes devas palielināšanas pētījumā smēķējošiem NSCLC pacientiem līdzsvara stāvokļa farmakokinētikas analīzes liecināja par devai proporcionālu erlotiniba iedarbības palielināšanos, palielinot Tarceva devu no 150 mg līdz maksimālajai pieļaujamajai devai-300. Šajā pētījumā plazmas koncentrācija smēķētājiem, lietojot 300 mg devu, bija 1,22 μg / ml (n = 17).
Pamatojoties uz farmakokinētikas pētījumu rezultātiem, pašreizējiem smēķētājiem jāiesaka Tarceva lietošanas laikā pārtraukt smēķēšanu, jo pretējā gadījumā koncentrācija plazmā var samazināties.
Pamatojoties uz populācijas farmakokinētikas analīzi, šķiet, ka opioīdu klātbūtne palielina iedarbību par aptuveni 11%.
Tika veikta otrā populācijas farmakokinētiskā analīze, ieskaitot erlotiniba datus no 204 aizkuņģa dziedzera vēža slimniekiem, kuri tika ārstēti ar erlotinibu un gemcitabīnu. Šī analīze parādīja, ka kovariāti, kas ietekmē erlotiniba klīrensu pētījuma pacientiem ar aizkuņģa dziedzera vēzi, bija ļoti līdzīgi tiem, kas tika novēroti iepriekšējā monoterapijas farmakokinētiskajā analīzē. Netika atklāta jauna kovariāta ietekme. Vienlaicīga gemcitabīna lietošana neietekmēja erlotiniba plazmas klīrensu.
Pediatriskā populācija: Nav veikti īpaši pētījumi ar pediatriskiem pacientiem
Gados vecāki pacienti: Nav veikti īpaši pētījumi ar gados vecākiem pacientiem.
Aknu mazspēja: erlotiniba klīrenss pārsvarā ir aknās. Pacientiem ar cietiem audzējiem un vidēji smagiem aknu darbības traucējumiem (Child-Pugh rādītājs 7–9) erlotiniba vidējais ģeometriskais AUC0-t un Cmax bija 27 000 ng • h / ml un 805 ng / ml, bet tie bija 29300 ng • h / ml un 1090 ng / ml pacientiem ar atbilstošu aknu darbību, ieskaitot pacientus ar primāru aknu vēzi vai metastāzēm aknās. Lai gan Cmax bija statistiski nozīmīgi zemāks pacientiem ar mērenu aknu mazspēju, šī atšķirība netiek uzskatīta par klīniski nozīmīgu. smagu aknu darbības traucējumu ietekme uz erlotiniba farmakokinētiku. Populācijas farmakokinētikas analīzē kopējā bilirubīna koncentrācijas palielināšanās serumā ir saistīta ar erlotiniba klīrensa palēnināšanos.
Nieru mazspēja: Erlotiniba un tā metabolītu izdalīšanās caur nierēm nav nozīmīga, jo mazāk nekā 9% no vienas devas izdalās ar urīnu. Klīniski nozīmīga korelācija starp erlotiniba klīrensu un kreatinīna klīrensu, bet nav pieejami dati par pacientiem ar kreatinīna klīrensu
05.3 Preklīniskie drošības dati
Starp sekām, kas novērotas pēc hroniskas ievadīšanas vismaz vienā dzīvnieku sugā vai pētījumā, ir ietekme uz radzeni (atrofija, čūlas), uz ādas (folikulu deģenerācija un iekaisums, apsārtums un alopēcija), uz olnīcām (atrofija), uz aknām (aknu nekroze), uz nierēm (nieru papilāru nekroze un cauruļveida paplašināšanās) un uz kuņģa -zarnu trakta (aizkavēta kuņģa iztukšošanās un caureja). Tika samazināti sarkano asins šūnu parametri un palielināts balto asins šūnu skaits, īpaši bija saistīta ALAT, ASAT un bilirubīna līmeņa paaugstināšanās, un šie dati tika iegūti par iedarbību, kas bija krietni zemāka par klīniski nozīmīgo.
Pamatojoties uz darbības mehānismu, erlotinibs ir potenciāli teratogēns. Dati no reproduktīvās toksicitātes pētījumiem ar žurkām un trušiem, lietojot devas, kas ir tuvu maksimālajai panesamajai devai, un / vai toksiskas mātītes devas norādīja uz reproduktīvo toksicitāti (žurku embriotoksicitāti), embriju rezorbciju un fetotoksicitāti. trušiem) un attīstību (samazināta mazuļu augšana un izdzīvošana žurkām), bet neuzrādīja teratogenitāti vai auglības traucējumus. Šie rezultāti tika novēroti klīniski nozīmīgai iedarbībai.
Parastie erlotiniba genotoksicitātes pētījumi ir bijuši negatīvi. Divu gadu kancerogenitātes pētījumi, kas veikti ar žurkām un pelēm, ar erlotinibu līdz koncentrācijai, kas pārsniedz terapeitisko koncentrāciju, kas izmantota cilvēkiem (attiecīgi līdz 2 un 10 reizes, pamatojoties uz Cmax un / vai AUC), bija negatīvi.
Žurkām pēc UV apstarošanas tika novērota viegla fototoksiska ādas reakcija.
06.0 FARMACEITISKĀ INFORMĀCIJA
06.1 Palīgvielas
Planšetdatora kodols:
Laktozes monohidrāts
Mikrokristāliskā celuloze (E460)
A tipa nātrija cietes glikolāts
Nātrija laurilsulfāts
Magnija stearāts (E470 b)
Tabletes pārklājums:
Hidroksipropilceluloze (E463)
Titāna dioksīds (E171)
Makrogols
Hipromeloze (E464)
06.2 Nesaderība
Nav būtisks.
06.3 Derīguma termiņš
4 gadi.
06.4 Īpaši uzglabāšanas nosacījumi
Šīm zālēm nav nepieciešami īpaši uzglabāšanas apstākļi.
06.5 Tiešā iepakojuma veids un iepakojuma saturs
PVC blisteris ar alumīnija foliju, kas satur 30 tabletes.
06.6 Norādījumi lietošanai un lietošanai
Nav īpašu norādījumu par iznīcināšanu.
Neizlietotās zāles un šo zāļu atkritumi jāiznīcina saskaņā ar vietējiem noteikumiem.
07.0 REĢISTRĀCIJAS APLIECĪBAS ĪPAŠNIEKS
Roche Registration Limited
6 Falcon Way
Šīras parks
Welwyn Garden City
AL7 1TW
Lielbritānija
08.0 REĢISTRĀCIJAS APLIECĪBAS NUMURS
EU/1/05/311/003
036871034
09.0 PIRMĀJAS APLIECĪBAS VAI ATĻAUJAS DATUMS
Reģistrācijas datums: 2005. gada 19. septembris
Pēdējās pārreģistrācijas datums: 2010. gada 19. septembris
10.0 TEKSTA PĀRSKATĪŠANAS DATUMS
2016. gada janvāris


















.jpg)