Aktīvās sastāvdaļas: sorafenibs
Nexavar 200 mg apvalkotās tabletes
Kāpēc lieto Nexavar? Kam tas paredzēts?
Nexavar lieto hepatokarcinomas ārstēšanai.
Nexavar lieto arī nieru vēža (progresējošas nieru šūnu karcinomas) ārstēšanai, ja tas ir progresējošā stadijā un ja standarta terapija nav palīdzējusi to apturēt vai tiek uzskatīta par nepiemērotu.
Nexavar lieto vairogdziedzera vēža (diferencēta vairogdziedzera vēža) ārstēšanai.
Nexavar ir tā sauktais daudzkināzes inhibitors. Tas darbojas, palēninot vēža šūnu augšanas ātrumu un bloķējot asins piegādi, kas ļauj vēža šūnām augt.
Kontrindikācijas Kad Nexavar nedrīkst lietot
Nelietojiet Nexavar
- ja Jums ir alerģija pret sorafenibu vai kādu citu (6. punktā minēto) šo zāļu sastāvdaļu.
Piesardzība lietošanā Kas jāzina pirms Nexavar lietošanas
Pirms Nexavar lietošanas konsultējieties ar ārstu vai farmaceitu.
Īpaša piesardzība, lietojot Nexavar, nepieciešama šādos gadījumos
- Ja rodas ādas problēmas. Nexavar var izraisīt izsitumus un ādas reakcijas, īpaši uz rokām un kājām. Šīs sekas parasti var ārstēt ārsts. Pretējā gadījumā ārsts var pārtraukt ārstēšanu vai pilnībā to pārtraukt.
- Ja Jums ir augsts asinsspiediens. Nexavar var izraisīt asinsspiediena paaugstināšanos; ārsts regulāri pārbaudīs asinsspiedienu un var izrakstīt zāles augsta asinsspiediena ārstēšanai.
- Ja Jums ir asiņošanas problēmas vai ja lietojat varfarīnu vai fenprokomonu. Ārstēšana ar Nexavar var palielināt asiņošanas risku. Ja lietojat varfarīnu vai fenprokomonu, zāles, kas atšķaida asinis, lai novērstu asins recekļu veidošanos, var būt paaugstināts asiņošanas risks.
- Ja Jums ir sāpes krūtīs vai sirdsdarbības traucējumi. Ārsts var izlemt pārtraukt ārstēšanu vai to pilnībā.
- Ja Jums ir sirdsdarbības traucējumi, piemēram, “elektriskā signāla traucējumi, ko sauc par“ QT pagarināšanos ”.
- Ja Jums ir vai ir tikko veikta operācija. Nexavar var ietekmēt brūču dzīšanu. Ja Jums ir paredzēta operācija, ārstēšana ar Nexavar, iespējams, tiks pārtraukta. Pēc tam ārsts izlems, kad to ņemt atpakaļ.
- Ja Jūs ārstējat ar irinotekānu vai docetakselu, kas arī ir zāles pret vēzi, Nexavar var pastiprināt šo zāļu iedarbību un jo īpaši blakusparādības.
- Ja lietojat neomicīnu vai citas antibiotikas. Nexavar efektivitāte var samazināties - ja Jums ir smaga aknu mazspēja, lietojot šīs zāles, Jums var būt saasinātas blakusparādības.
- Ja Jums ir pavājināta nieru darbība. Ārsts uzraudzīs ūdens un elektrolītu līdzsvaru.
- Auglība. Nexavar var samazināt auglību gan vīriešiem, gan sievietēm. Ja tas attiecas uz jums, konsultējieties ar ārstu.
- Ārstēšanas laikā var rasties kuņģa -zarnu trakta perforācija (skatīt 4. sadaļu: Iespējamās blakusparādības). Šajā gadījumā ārsts pārtrauc ārstēšanu.
- Ja Jums ir vairogdziedzera vēzis, ārsts pārbaudīs kalcija un vairogdziedzera hormona līmeni asinīs.
Pastāstiet ārstam, ja kāds no šiem gadījumiem attiecas uz Jums. Jums var būt nepieciešama šo problēmu ārstēšana, vai arī ārsts var mainīt Nexavar devu vai vispār pārtraukt ārstēšanu (skatīt arī 4. punktu: Iespējamās blakusparādības).
Bērni un pusaudži
Nexavar vēl nav pētīts bērniem un pusaudžiem.
Mijiedarbība Kādas zāles vai pārtikas produkti var mainīt Nexavar iedarbību
Dažas zāles var ietekmēt Nexavar vai tas var ietekmēt to. Pastāstiet ārstam vai farmaceitam par visām zālēm, kuras lietojat pēdējā laikā, esat lietojis vai varētu lietot, vai citas zāles, ieskaitot šīs zāles, ieskaitot zāles, ko var iegādāties bez receptes:
- Rifampicīns, neomicīns vai citas zāles, ko lieto infekciju ārstēšanai (antibiotikas)
- Hypericum perforatum, pazīstams arī kā "asinszāle", augu izcelsmes zāles depresijas ārstēšanai
- Fenitoīns, karbamazepīns vai fenobarbitāls, epilepsijas un citu slimību ārstēšana
- Deksametazons, kortikosteroīds, ko lieto dažādām slimībām
- Varfarīns vai fenprokomons, antikoagulanti, ko lieto, lai novērstu asins recekļu veidošanos
- Doksorubicīns, kapecitabīns, docetaksels, paklitaksels un irinotekāns, ko lieto vēža ārstēšanā.
- Digoksīns, ko lieto vieglas vai vidēji smagas sirds mazspējas ārstēšanai
Brīdinājumi Ir svarīgi zināt, ka:
Grūtniecība un zīdīšanas periods
Nexavar terapijas laikā izvairieties no grūtniecības. Ja esat reproduktīvā vecumā, ārstēšanas laikā ar Nexavar jāizmanto efektīva kontracepcijas metode. Ja ārstēšanas laikā ar Nexavar Jums iestājas grūtniecība, nekavējoties pastāstiet savam ārstam, kurš izlems, vai ārstēšanu turpināt vai pārtraukt.
Ārstēšanas laikā ar Nexavar nedrīkst barot bērnu ar krūti, jo šīs zāles var traucēt mazuļa augšanu un attīstību.
Transportlīdzekļu vadīšana un mehānismu apkalpošana
Nav pamata uzskatīt, ka Nexavar ietekmēs spēju vadīt transportlīdzekļus vai apkalpot mehānismus.
Deva, lietošanas veids un laiks Kā lietot Nexavar: Devas
Ieteicamā Nexavar deva pieaugušajiem ir divas 200 mg tabletes divas reizes dienā.
Tās atbilst dienas devai 800 mg vai četrām tabletēm dienā. Lietojiet Nexavar tabletes, uzdzerot glāzi ūdens, starp ēdienreizēm vai kopā ar pārtiku ar zemu vai vidēju tauku saturu. Nelietojiet šīs zāles kopā ar ļoti taukainu pārtiku, jo tās var samazināt to efektivitāti. Ja plānojat ēst ļoti taukainu pārtiku, lietojiet tabletes vismaz 1 stundu pirms vai 2 stundas pēc pusdienām. Vienmēr lietojiet šīs zāles tieši tā, kā ārsts Jums stāstījis. Ja neesat pārliecināts, konsultējieties ar ārstu vai farmaceitu.
Ir svarīgi lietot šīs zāles katru dienu aptuveni vienā un tajā pašā laikā, lai koncentrācija asinīs būtu nemainīga.
Šīs zāles parasti lieto, kamēr tiek atzīmēts klīniskais ieguvums, un nav nepanesamu blakusparādību.
Pārdozēšana Ko darīt, ja esat lietojis pārāk daudz Nexavar
Ja esat lietojis Nexavar vairāk nekā noteikts
Nekavējoties pastāstiet ārstam, ja Jūs vai kāds cits esat lietojis vairāk nekā noteikts. Ja esat lietojis pārāk daudz Nexavar, blakusparādības, īpaši caureja un ādas reakcijas, kļūst iespējamas vai nopietnākas. Ārsts var ieteikt pārtraukt šo zāļu lietošanu.
Ja esat aizmirsis lietot Nexavar
Ja esat aizmirsis lietot devu, ieņemiet to, tiklīdz atceraties. Ja nākamajai devai pietrūkst laika, aizmirstiet aizmirsto devu un turpiniet ar biežumu
Blakusparādības Kādas ir Nexavar blakusparādības?
Tāpat kā citas zāles, šīs zāles var izraisīt blakusparādības, kaut arī ne visiem tās izpaužas. Šīs zāles var arī mainīt dažu asins analīžu rezultātus.
Ļoti bieži sastopams:
var skart vairāk nekā 1 no 10 cilvēkiem
- caureja
- savārgums (slikta dūša)
- vājuma vai noguruma sajūta (nogurums)
- sāpes (ieskaitot sāpes mutē, vēderā, galvassāpes, kaulu sāpes, vēža sāpes)
- matu izkrišana (alopēcija)
- apsārtums vai sāpes plaukstās vai pēdās (roku un kāju ādas reakcija)
- nieze vai izsitumi
- Viņš atrāvās
- asiņošana (ieskaitot asiņošanu smadzenēs, zarnu sienās un elpceļos)
- augsts asinsspiediens vai paaugstināts asinsspiediens (hipertensija)
- infekcijas
- apetītes zudums (anoreksija)
- aizcietējums
- locītavu sāpes (artralģija)
- drudzis
- svara zudums
- ādas sausums
Bieži:
var skart līdz 1 no 10 cilvēkiem
- gripai līdzīga slimība
- gremošanas traucējumi (dispepsija)
- rīšanas grūtības (disfāgija)
- iekaisums vai sausums mutē, sāpes mēlē (stomatīts un gļotādas iekaisums)
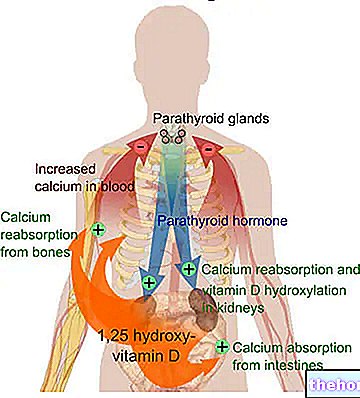
- zems kalcija līmenis asinīs (hipokalciēmija)
- zems kālija līmenis asinīs (hipokaliēmija)
- muskuļu sāpes (mialģija)
- pirkstu un kāju pirkstu jutīguma traucējumi, tai skaitā tirpšana un nejutīgums (perifēra maņu neiropātija)
- depresija
- erekcijas problēmas (impotence)
- balss izmaiņas (disfonija)
- pinnes
- iekaisusi, sausa vai zvīņojoša āda (dermatīts, ādas lobīšanās)
- sirdskaite
- sirdslēkme (miokarda infarkts) vai sāpes krūtīs
- troksnis ausīs (zvana ausīs)
- nieru mazspēja
- augsts olbaltumvielu līmenis urīnā (proteīnūrija)
- vispārējs vājums vai spēka zudums (astēnija)
- samazināts balto asins šūnu skaits (leikopēnija un neitropēnija)
- samazināts sarkano asins šūnu skaits (anēmija)
- zems trombocītu skaits asinīs (trombocitopēnija)
- matu folikulu iekaisums (folikulīts)
- samazināta vairogdziedzera darbība (hipotireoze)
- zems nātrija līmenis asinīs (hiponatriēmija)
- garšas sajūtas izmaiņas (disgeizija)
- sejas un bieži citu ādas zonu apsārtums (pietvīkums)
- iesnas (iesnas)
- grēmas (gastroezofageālā refluksa slimība)
- ādas vēzis (keratoakantoma / plakanšūnu ādas vēzis)
- ādas ārējā slāņa sabiezēšana (hiperkeratoze)
- pēkšņa patvaļīga muskuļu kontrakcija (muskuļu spazmas)
Retāk:
var skart līdz 1 no 100 cilvēkiem
- kuņģa iekaisums (gastrīts)
- kuņģa (vēdera) sāpes pankreatīta, žultspūšļa un / vai žultsvadu iekaisuma dēļ
- ādas vai acu dzelte (dzelte), ko izraisa augsts žults pigmentu līmenis (hiperbilirubinēmija)
- alerģiskas reakcijas (ieskaitot ādas reakcijas un nātreni)
- dehidratācija
- krūšu palielināšanās (ginekomastija)
- apgrūtināta elpošana (plaušu slimība)
- ekzēma
- pārmērīga vairogdziedzera darbība (hipertireoze)
- vairāki ādas izsitumi (multiformā eritēma)
- augsts asinsspiediens
- kuņģa -zarnu trakta perforācija
- atgriezeniska tūska smadzeņu aizmugurē, kas var būt saistīta ar galvassāpēm, apziņas izmaiņām, krampjiem un redzes simptomiem, ieskaitot redzes zudumu (atgriezeniska atgriezeniska leikoencefalopātija)
- pēkšņa, smaga alerģiska reakcija (anafilaktiska reakcija)
Reti:
var skart līdz 1 no 1000 cilvēkiem
- alerģiska reakcija ar ādas (piemēram, sejas, mēles) pietūkumu, kas var apgrūtināt elpošanu un rīšanu (angioneirotiskā tūska)
- patoloģisks sirds ritms (QT pagarināšanās)
- Aknu iekaisums, kas var izraisīt sliktu dūšu, vemšanu, sāpes vēderā un dzelti (zāļu izraisīts hepatīts)
- "saules apdegumiem līdzīgi izsitumi uz ādas, kas iepriekš bijusi pakļauta staru terapijai un var būt smaga (aktīniskam līdzīgs dermatīts)
- smagas ādas un / vai gļotādu reakcijas, kas var ietvert sāpīgus pūslīšus un drudzi, atdaloties lielām ādas vietām (Stīvensa-Džonsona sindroms un toksiska epidermas nekrolīze)
- patoloģiski muskuļu bojājumi, kas var izraisīt nieru darbības traucējumus (rabdomiolīze)
- nieru bojājumi, kas izraisa lielu daudzumu olbaltumvielu urīnā (nefrotiskais sindroms)
- ādas asinsvadu iekaisums, kas var izpausties kā izsitumi (leikocitoklastiskais vaskulīts)
Nezinams:
biežumu nevar noteikt pēc pieejamajiem datiem
- smadzeņu darbības traucējumi, kas var būt saistīti, piemēram, ar miegainību, uzvedības izmaiņām vai apjukumu (encefalopātija)
Ziņošana par blakusparādībām
Ja Jums rodas jebkādas blakusparādības, konsultējieties ar ārstu vai farmaceitu. Tas attiecas arī uz iespējamām blakusparādībām, kas nav minētas šajā instrukcijā. Jūs varat ziņot par blakusparādībām arī tieši, izmantojot V pielikumā minēto valsts ziņošanas sistēmu. Ziņojot par blakusparādībām, jūs varat palīdzēt iegūt vairāk informācijas par šo zāļu drošumu.
Derīguma termiņš un saglabāšana
Uzglabāt šīs zāles bērniem neredzamā un nepieejamā vietā.
Nelietot šīs zāles pēc derīguma termiņa beigām, kas norādīts uz kastītes pēc “Derīgs līdz:” un uz katra blistera pēc “Derīgs līdz”. Derīguma termiņš attiecas uz norādītā mēneša pēdējo dienu.
Uzglabāt šīs zāles temperatūrā līdz 25 ° C.
Neizmetiet zāles kanalizācijā vai sadzīves atkritumos. Jautājiet farmaceitam, kā izmest zāles, kuras vairs nelietojat. Tas palīdzēs aizsargāt vidi.
Ko Nexavar satur
- Aktīvā viela ir sorafenibs. Katra apvalkotā tablete satur 200 mg sorafeniba (tozilāta veidā).
- Citas sastāvdaļas ir: Tabletes kodols: nātrija kroskarmeloze, mikrokristāliskā celuloze, hipromeloze, nātrija laurilsulfāts un magnija stearāts. Tabletes apvalks: hipromeloze, makrogols, titāna dioksīds (E 171) un sarkanais dzelzs oksīds (E 172)
Nexavar ārējais izskats un iepakojums
Nexavar 200 mg apvalkotās tabletes ir sarkanas un apaļas, ar Bayer krustiņu vienā pusē un "200" otrā pusē. Tās ir iepakotas 112 tablešu kastītēs, kas satur četrus caurspīdīgus kalendāra blisterus pa 28 tabletēm katrā.
Avota lietošanas instrukcija: AIFA (Itālijas zāļu aģentūra). Saturs publicēts 2016. gada janvārī. Pašlaik pieejamā informācija var nebūt atjaunināta.
Lai piekļūtu visjaunākajai versijai, ieteicams piekļūt AIFA (Itālijas zāļu aģentūra) vietnei. Atruna un noderīga informācija.
01.0 ZĀĻU NOSAUKUMS
NEXAVAR 200 MG
02.0 KVALITATĪVAIS UN KVANTITATĪVAIS SASTĀVS
Katra apvalkotā tablete satur 200 mg sorafeniba (tozilāta veidā).
Pilnu palīgvielu sarakstu skatīt apakšpunktā 6.1.
03.0 ZĀĻU FORMA
Apvalkotā tablete (tablete).
Sarkanas, apaļas, abpusēji izliektas apvalkotās tabletes ar marķējumu ar Bayer krustu vienā pusē un "200" otrā.
04.0 KLĪNISKĀ INFORMĀCIJA
04.1 Terapeitiskās indikācijas
Hepatokarcinoma
Nexavar ir indicēts aknu šūnu karcinomas ārstēšanai (skatīt apakšpunktu 5.1).
Nieru šūnu karcinoma
Nexavar ir indicēts pacientu ar progresējošu nieru šūnu karcinomu ārstēšanai, kuriem iepriekšējā alfa-interferona vai interleikīna-2 terapija nav bijusi veiksmīga vai kuri tiek uzskatīti par nepiemērotiem šādai terapijai.
Diferencēts vairogdziedzera vēzis
Nexavar ir indicēts pacientiem ar lokāli progresējošu vai metastātisku, progresējošu, ar radioaktīvo jodu neārstējamu diferencētu vairogdziedzera vēzi (papilāru / folikulāru / Hirtla šūnu).
04.2 Devas un lietošanas veids
Ārstēšana ar Nexavar jāveic ārsta uzraudzībā, kuram ir pieredze pretvēža terapijas lietošanā.
Devas
Ieteicamā Nexavar deva pieaugušajiem ir 400 mg sorafeniba (divas 200 mg tabletes) divas reizes dienā (līdzvērtīga kopējai dienas devai 800 mg).
Ārstēšana jāturpina tik ilgi, kamēr tiek novērots klīniskais ieguvums, vai līdz parādās nepieņemama toksicitāte.
Devas pielāgošana
Lai ārstētu iespējamās zāļu blakusparādības, var būt nepieciešams īslaicīgi pārtraukt vai samazināt sorafeniba terapiju.
Ja hepatocelulārās karcinomas ārstēšanas laikā nepieciešama devas samazināšana (aknu šūnu karcinoma, HCC) un nieru šūnu karcinomu (nieru šūnu karcinoma(RCC)), Nexavar deva jāsamazina līdz divām 200 mg sorafeniba tabletēm vienu reizi dienā (skatīt apakšpunktu 4.4).
Ja diferencēta vairogdziedzera vēža ārstēšanas laikā nepieciešama devas samazināšana (diferencēta vairogdziedzera karcinoma(DTC), Nexavar deva jāsamazina līdz 600 mg sorafeniba dienā, sadalot devās (divas 200 mg tabletes un viena 200 mg tablete ar divpadsmit stundu intervālu).
Ja nepieciešama turpmāka devas samazināšana, Nexavar var samazināt līdz 400 mg sorafeniba dienā, sadalot devās (divas 200 mg tabletes ar divpadsmit stundu intervālu) un, ja nepieciešams, vēl vairāk samazināt līdz vienai 200 mg tabletei vienu reizi dienā. blakusparādības, Nexavar devu var palielināt.
Pediatriskā populācija
Nexavar drošība un efektivitāte vecākiem bērniem un pusaudžiem
Vecāka gadagājuma cilvēki
Gados vecākiem cilvēkiem (pacientiem, kas vecāki par 65 gadiem) devas pielāgošana nav nepieciešama.
Nieru darbības traucējumi
Pacientiem ar viegliem, vidēji smagiem vai smagiem nieru darbības traucējumiem devas pielāgošana nav nepieciešama. Dati par pacientiem, kuriem tiek veikta dialīze, nav pieejami (skatīt 5.2. Apakšpunktu).
Pacientiem ar nieru mazspējas risku ieteicams kontrolēt ūdens un elektrolītu līdzsvaru.
Aknu darbības traucējumi
Pacientiem ar Child Pugh A vai B (viegliem līdz vidēji smagiem) aknu darbības traucējumiem deva nav jāpielāgo. Nav pieejami dati par pacientiem ar smagiem Child Pugh C aknu darbības traucējumiem (skatīt 4.4. Un 5.2. Apakšpunktu).
Lietošanas veids
Iekšķīgai lietošanai
Sorafenibs jāievada starp ēdienreizēm vai maltīti ar zemu vai mērenu tauku saturu. Ja pacients plāno ieturēt maltīti ar augstu tauku saturu, sorafeniba tabletes jālieto vismaz vienu stundu pirms vai divas stundas pēc ēšanas. Tabletes jānorij, uzdzerot glāzi ūdens.
04.3 Kontrindikācijas
Paaugstināta jutība pret aktīvo vielu vai jebkuru no 6.1. Apakšpunktā uzskaitītajām palīgvielām.
04.4 Īpaši brīdinājumi un piesardzība lietošanā
Dermatoloģiskā toksicitāte
Roku un pēdu ādas reakcija (plaukstas-plantāra eritrodizestēzija) e izsitumi ir visbiežāk sastopamās sorafeniba blakusparādības. Izsitumi un roku un kāju ādas reakcija parasti ir 1. un 2. pakāpe, saskaņā ar i Kopējie toksicitātes kritēriji (CTC) un parasti parādās sorafeniba terapijas pirmo sešu nedēļu laikā. Dermatoloģiskās toksicitātes ārstēšana var ietvert lokālu terapiju, lai mazinātu simptomus, īslaicīgu ārstēšanas pārtraukšanu un / vai sorafeniba devas maiņu, vai smagos vai pastāvīgos gadījumos galīgu zāļu lietošanas pārtraukšanu (skatīt 4.8. Apakšpunktu).
Hipertensija
Pacientiem, kuri tika ārstēti ar sorafenibu, tika novērota lielāka arteriālās hipertensijas sastopamība.Šiem pacientiem hipertensija parasti bija viegla vai vidēji smaga, parādījās ārstēšanas sākumposmā un reaģēja uz standarta antihipertensīvo terapiju. Regulāri jākontrolē asinsspiediens un pēc vajadzības jāārstē saskaņā ar pašreizējo medicīnas praksi. Smagas vai pastāvīgas hipertensijas vai hipertensīvas krīzes gadījumā, neraugoties uz antihipertensīvās terapijas uzsākšanu, ieteicams apsvērt iespēju neatgriezeniski pārtraukt sorafeniba lietošanu (skatīt apakšpunktu 4.8).
Asiņošana
Pēc sorafeniba lietošanas var palielināties asiņošanas risks. Ja asiņošanas epizodei nepieciešama medicīniska iejaukšanās, ieteicams apsvērt neatgriezenisku sorafeniba lietošanas pārtraukšanu (skatīt apakšpunktu 4.8).
Sirds išēmija un / vai sirdslēkme
Dubultmaskētā, randomizētā, placebo kontrolētā pētījumā (1. pētījums, skatīt 5.1. Apakšpunktu) ārstēšanas sākuma sirds infarkta vai išēmijas sastopamība bija augstāka sorafeniba grupā (4,9%) nekā ārstēšanās grupā. 3. pētījumā (skatīt 5.1. Apakšpunktu) ar sirds infarktu vai išēmiju, kas sākās ar ārstēšanu, sastopamības biežums bija 2,7% ar sorafenibu ārstētiem pacientiem un 1,3% ar placebo ārstētiem pacientiem. No šiem pētījumiem tika izslēgti pacienti ar nestabilu koronāro artēriju slimību vai nesenu miokarda infarktu. Pacientiem, kuriem attīstās sirds išēmija un / vai infarkts, jāapsver nepieciešamība uz laiku vai pastāvīgi pārtraukt ārstēšanu ar sorafenibu (skatīt apakšpunktu 4.8).
QT intervāla pagarinājums
Ir pierādīts, ka sorafenibs pagarina QT / QTc intervālu (skatīt 5.1. Apakšpunktu), kas var palielināt sirds kambaru aritmijas risku. Lietojiet sorafenibu piesardzīgi pacientiem, kuriem ir vai var attīstīties QTc intervāla pagarināšanās, piemēram, pacientiem ar iedzimtu garu QT Sindroms, pacienti, kuri tiek ārstēti ar lielu kumulatīvo antraciklīnu devu, pacienti, kuri lieto noteiktus antiaritmiskus līdzekļus vai citas zāles, kas var izraisīt QT intervāla pagarināšanos, un pacienti ar elektrolītu traucējumiem, piemēram, hipokaliēmiju, hipokalciēmiju vai hipomagnēmiju Ja šiem pacientiem lieto sorafenibu, periodiska elektrokardiogrāfija un ārstēšanas laikā jāveic elektrolītu (magnija, kālija un kalcija) mērījumi.
Kuņģa -zarnu trakta perforācija
Kuņģa -zarnu trakta perforācija ir retums, un par to ziņots mazāk nekā 1% pacientu, kuri lieto sorafenibu. Dažos gadījumos nebija saistības ar acīmredzamu intraabdominālu audzēju. Kuņģa -zarnu trakta perforācijas gadījumā sorafeniba lietošana jāpārtrauc (skatīt apakšpunktu 4.8).
Aknu darbības traucējumi
Nav pieejami dati par pacientiem ar smagiem aknu darbības traucējumiem (Child Pugh C). Šādiem pacientiem iedarbība var palielināties, jo sorafenibs tiek izvadīts galvenokārt caur aknām (skatīt 4.2. Un 5.2. Apakšpunktu).
Vienlaicīga varfarīna lietošana
Retas asiņošanas epizodes vai INR palielināšanās (Starptautiski normalizēts
Attiecība) ir ziņots par dažiem pacientiem, kuri sorafeniba terapijas laikā lietoja varfarīnu. Pacienti, kuri saņem varfarīna vai fenprokumona terapiju, regulāri jānovēro, lai noteiktu protrombīna laika izmaiņas, INR vai klīniski nozīmīgas asiņošanas epizodes (skatīt 4.5. Un 4.8. Apakšpunktu).
Komplikācijas brūču dzīšanā
Nav veikti oficiāli pētījumi par sorafeniba ietekmi uz brūču dzīšanu. Pacientiem, kuriem tiek veikta liela operācija, piesardzības nolūkos ieteicams īslaicīgi pārtraukt ārstēšanu ar sorafenibu. Klīniskā pieredze par to, kad atsākt terapiju pēc lielas operācijas, ir ierobežota. Tādēļ lēmums atsākt sorafeniba terapiju pēc lielas operācijas jāpamato ar klīnisku novērtējumu par atbilstošu brūču sadzīšanu.
Vecāka gadagājuma cilvēki
Ir ziņots par nieru mazspējas gadījumiem. Tādēļ jāapsver nieru darbības uzraudzība.
Mijiedarbība starp narkotikām
Ieteicams ievērot piesardzību, lietojot sorafenibu kopā ar vielām, kas galvenokārt tiek metabolizētas un / vai izvadītas caur UGT1A1 (piemēram, irinotekānu) vai UGT1A9 (skatīt apakšpunktu 4.5).
Ieteicama piesardzība, ja vienlaikus tiek lietots sorafenibs un docetaksels (skatīt 4.5. Apakšpunktu).
Kombinācija ar neomicīnu vai citām antibiotikām, kas var izraisīt nopietnus ekoloģiskus traucējumus kuņģa -zarnu trakta mikroflorā, var izraisīt sorafeniba bioloģiskās pieejamības samazināšanos (skatīt 4.5. Apakšpunktu). Pirms terapijas uzsākšanas jānovērtē sorafeniba koncentrācijas samazināšanās risks plazmā. ārstēšanas kurss ar antibiotikām.
Paaugstināta mirstība tika novērota pacientiem ar plakanšūnu plaušu vēzi, kuri tika ārstēti ar sorafenibu kombinācijā ar ķīmijterapiju uz platīna bāzes.
Divos randomizētos klīniskos pētījumos, kuros pētīja pacientus ar nesīkšūnu plaušu vēzi (Nesīkšūnu plaušu vēzis(NSCLC), riska pakāpe (HR) kopējai dzīvildzei ar plakanšūnu plaušu vēzi pacientu apakšgrupā bija 1,81 (95% TI 1,19, 2,74) pacientiem, kuri tika ārstēti ar sorafenibu papildus paklitaksela / karboplatīna terapijai, un 1,22 (95% CI 0,82; 1,80) pacientiem, kuri tika ārstēti ar sorafenibu papildus gemcitabīna / cisplatīna terapijai. Galvenais nāves cēlonis netika novērots, bet pacientiem, kuri tika ārstēti ar sorafenibu papildus terapijai, kas balstīta uz platīnu, tika novērots palielināts elpošanas mazspējas, asiņošanas un infekcijas gadījumu skaits.
Patoloģijas brīdinājumi
Diferencēta vairogdziedzera karcinoma (DTC)
Pirms ārstēšanas uzsākšanas ārstiem ieteicams rūpīgi izvērtēt individuālo pacienta prognozi, pamatojoties uz maksimālo bojājuma lielumu (skatīt 5.1. Apakšpunktu), ar slimību saistītos simptomus (skatīt 5.1. Apakšpunktu) un progresēšanas ātrumu.
Lai ārstētu iespējamās zāļu blakusparādības, var būt nepieciešams "īslaicīgi pārtraukt vai samazināt sorafeniba terapiju. 5. pētījumā (skatīt 5.1. Apakšpunktu) 37% pacientu uz laiku pārtrauca terapiju un 35% samazināja devu jau 1. sorafeniba terapijas ciklā.
Devas samazināšana bija tikai daļēji efektīva, lai mazinātu nevēlamās blakusparādības, tāpēc ieteicams atkārtoti novērtēt ieguvumus un risku, ņemot vērā pretvēža darbību un panesamību.
Asiņošana DTC
Iespējamā asiņošanas riska dēļ pirms sorafeniba ievadīšanas pacientiem ar DTC trahejas, bronhu un barības vada infiltrācija jāārstē ar lokālu terapiju.
Hipokalciēmija DTC
Lietojot sorafenibu pacientiem ar DTC, ieteicams rūpīgi uzraudzīt kalcija līmeni asinīs. Klīniskajos pētījumos hipokalciēmija bija biežāka un smagāka pacientiem ar DTC, īpaši tiem, kuriem anamnēzē bija hipoparatireoze, salīdzinot ar pacientiem ar nieru šūnu karcinomu vai hepatokarcinomu. 3. un 4. pakāpes hipokalciēmija parādījās 6,8% un 3,4% ar sorafenibu ārstētiem pacientiem ar DTC (skatīt apakšpunktu 4.8). Smaga hipokalciēmija ir jākoriģē, lai novērstu tādas komplikācijas kā QT intervāla pagarināšana vai torsades de pointes (skatīt sadaļu QT intervāla pagarināšana).
TSH nomākšana DTC
5. pētījumā (skatīt 5.1. Apakšpunktu) ar sorafenibu ārstētiem pacientiem tika novērota TSH līmeņa paaugstināšanās vairāk nekā 0,5 mU / l. Lietojot sorafenibu pacientiem ar DTC, ieteicams rūpīgi kontrolēt TSH līmeni.
Nieru šūnu karcinoma
Paaugstināta riska pacienti, kā definējusi MSKCC prognostiskā grupa (Slouna Ketteringa memoriālais vēža centrs), nebija iekļauti III fāzes klīniskajā pētījumā ar nieru šūnu karcinomu (skatīt 1. pētījumu 5.1. apakšpunktā), un ieguvuma un riska attiecība šiem pacientiem nav noteikta.
04.5 Mijiedarbība ar citām zālēm un citi mijiedarbības veidi
Metabolisma enzīmu induktori
Lietojot rifampicīnu 5 dienas pirms vienas sorafeniba devas ievadīšanas, vidējais sorafeniba AUC samazinājās par 37%. Citi CYP3A4 un / vai glikuronidācijas induktori (piem. hypericum perforatum pazīstams arī kā "asinszāle", fenitoīns, karbamazepīns, fenobarbitāls un deksametazons) var paātrināt sorafeniba metabolismu un tādējādi samazināt tā koncentrāciju.
CYP3A4 inhibitori
Ketokonazols, spēcīgs CYP3A4 inhibitors, ievadīts vienu reizi dienā 7 dienas veseliem brīvprātīgajiem vīriešiem, nemainīja vienas 50 mg sorafeniba devas vidējo AUC. Šie dati liecina, ka sorafeniba un CYP3A4 inhibitoru klīniskā farmakokinētiskā mijiedarbība ir maz ticama.
CYP2B6, CYP2C8 un CYP2C9 substrāti
In vitro sorafenibs gandrīz vienādi spēcīgi inhibē CYP2B6, CYP2C8 un CYP2C9. Tomēr klīniskajos farmakokinētikas pētījumos 400 mg sorafeniba lietošana divas reizes dienā ar ciklofosfamīdu, CYP2B6 substrātu, vai paklitaksela, kas ir CYP2C8 substrāts, neizraisīja klīniski nozīmīgu inhibīciju. Šie dati liecina, ka sorafenibs ieteicamajā 400 devā mg divas reizes dienā, var nebūt inhibitors in vivo CYP2B6 vai CYP2C8.
Turklāt vienlaicīga ārstēšana ar sorafenibu un varfarīnu, kas ir CYP2C9 substrāts, neizraisīja izmaiņas vidējā PT-INR salīdzinājumā ar placebo. Tāpēc arī "inhibīcijas" risks in vivo klīniski nozīmīgu sorafeniba CYP2C9 var uzskatīt par zemu. Tomēr pacientiem, kuri lieto varfarīnu vai fenprokumonu, regulāri jākontrolē INR (skatīt apakšpunktu 4.4).
CYP3A4, CYP2D6 un CYP2C19 substrāti
Vienlaicīga sorafeniba un midazolāma, dekstrometorfāna vai omeprazola, kas ir attiecīgi citohromu CYP3A4, CYP2D6 un CYP2C19 substrāti, lietošana nemainīja šo līdzekļu iedarbību. Tas norāda, ka sorafenibs nav ne šo izoenzīmu inhibitors, ne induktors. citohroms P450, tāpēc sorafeniba klīniskā farmakokinētiskā mijiedarbība ar šo enzīmu substrātiem ir maz ticama.
UGT1A1 un UGT1A9 substrāti
In vitro, sorafenibs inhibēja glikuronidāciju, ko izraisīja UGT1A1 un UGT1A9. Šī konstatējuma klīniskā nozīme nav zināma (skatīt zemāk un 4.4. Apakšpunktu).
Izglītība in vitro par CYP sistēmas enzīmu indukciju
Pēc sorafeniba iedarbības uz cilvēka hepatocītu kultūrām CYP1A2 un CYP3A4 aktivitātes nemainījās, tādējādi norādot, ka sorafenibs, visticamāk, nebūs CYP1A2 un CYP3A4 induktors.
Pamatnes P-gp
In vitroIr pierādīts, ka sorafenibs inhibē transporta proteīna p-glikoproteīnu (P-gp). Vienlaicīgas ārstēšanas ar sorafenibu gadījumā nevar izslēgt P-gp substrātu, piemēram, digoksīna, koncentrācijas palielināšanos plazmā.
Asociācija ar citiem pretvēža līdzekļiem
Klīniskajos pētījumos sorafenibu lietoja kopā ar vairākiem citiem pretaudzēju līdzekļiem atbilstoši to parastajai devai, ieskaitot gemcitabīnu, cisplatīnu, oksaliplatīnu, paklitakselu, karboplatīnu, kapecitabīnu, doksorubicīnu, irinotekānu, docetakselu un ciklofosfamīdu. Sorafenibam nebija klīniski nozīmīgas ietekmes uz gemcitabīna, cisplatīna, karboplatīna, oksaliplatīna vai ciklofosfamīda farmakokinētiku.
Paklitaksels / karboplatīns
• paklitaksela (225 mg / m2) un karboplatīna (AUC = 6) lietošana kopā ar sorafenibu (≤ 400 mg divas reizes dienā), ar 3 dienu pārtraukumu sorafeniba ievadīšanai (iepriekšējās divas dienas un paklitaksela / karboplatīna lietošanas dienu) ), neietekmēja paklitaksela farmakokinētiku.
• Paklitaksela (225 mg / m palielinājās paklitaksela iedarbība un par 50% palielinājās 6-OH paklitaksela iedarbība.Karboplatīna farmakokinētika netika ietekmēta.
Šie dati liecina, ka devas pielāgošana nav nepieciešama, ja paklitakselu un karboplatīnu lieto vienlaikus ar sorafenibu, 3 dienas pārtraucot sorafeniba ievadīšanu (divas dienas pirms un paklitaksela / karboplatīna ievadīšanas dienas). Ņemiet vērā palielināta sorafeniba un paklitaksela iedarbība drīz pēc vienlaicīgas sorafeniba lietošanas, nepārtraucot zāļu lietošanu.
Kapecitabīns
Vienlaicīga kapecitabīna (750–1050 mg / m2 divas reizes dienā, 1. – 14. Diena ik pēc 21 dienas) un sorafeniba (200 vai 400 mg divas reizes dienā, nepārtraucot devu) lietošana vienlaicīgi būtiski neizmainīja sorafeniba iedarbību, bet Kapecitabīna iedarbība palielinās par -50% un 5-FU iedarbība palielinās par 0-52%. Šo nelielo, pieticīgo 5-FU iedarbības pieaugumu klīniskā nozīme nav zināma.
Doksorubicīns / irinotekāns
Vienlaicīga ārstēšana ar sorafenibu izraisīja doksorubicīna AUC palielināšanos par 21%. Lietojot kopā ar irinotekānu, kura metabolīts SN-38 pēc tam tiek metabolizēts caur UGT1A1 ceļu, SN-38 AUC palielinājās par 67-120% un 26 - 42% irinotekāna AUC. " Šo datu klīniskā nozīme nav zināma (skatīt apakšpunktu 4.4).
Docetaksels
Docetaksels (deva 75 vai 100 mg / m2 ik pēc 21 dienas), ko lieto vienlaikus ar sorafenibu (200 mg vai 400 mg divas reizes dienā 21 dienas terapijas kursa 2. līdz 19. dienā, ar 3 dienu pārtraukumu, kas atbilst docetakselam docetaksela AUC un Cmax palielinājās attiecīgi par 36 - 80% un 16 - 32% .. Lietojot vienlaikus ar sorafenibu un docetakselu, ieteicams ievērot piesardzību (skatīt apakšpunktu 4.4).
Asociācija ar citiem aģentiem
Neomicīns
Kombinācija ar neomicīnu-nesistēmisku pretmikrobu līdzekli, ko izmanto kuņģa-zarnu trakta floras iznīcināšanai, traucē sorafeniba enterohepatisko recirkulāciju (skatīt 5.2. Apakšpunktu, Biotransformācija un metabolisms), kā rezultātā samazinās sorafeniba iedarbība. Veseliem brīvprātīgajiem, kuri tika ārstēti ar neomicīnu 5 dienas vidējā sorafeniba iedarbība samazinājās par 54%. Citu antibiotiku iedarbība nav pētīta, bet, visticamāk, būs atkarīga no to spējas traucēt mikroorganismiem ar glikuronidāzes aktivitāti.
04.6 Grūtniecība un zīdīšana
Grūtniecība
Nav datu par sorafeniba lietošanu grūtniecēm. Pētījumos ar dzīvniekiem ir pierādīta toksiska ietekme uz reproduktīvo funkciju, tostarp malformācijas (skatīt 5.3. Apakšpunktu). Ir pierādīts, ka sorafenibs un tā metabolīti šķērso placentu žurkām. Sorafenibu nedrīkst lietot grūtniecības laikā, ja vien tas nav nepārprotami nepieciešams, un tikai pēc rūpīgas mātes vajadzību un augļa riska apsvēršanas.
Sievietēm reproduktīvā vecumā ārstēšanas laikā jāizmanto efektīva kontracepcijas metode.
Barošanas laiks
Nav zināms, vai sorafenibs izdalās mātes pienā. Dzīvniekiem sorafenibs un / vai tā metabolīti izdalās pienā.Tā kā sorafenibs var pasliktināt jaundzimušā augšanu un attīstību (skatīt 5.3. Apakšpunktu), ārstēšanas laikā ar sorafenibu sievietēm jāpārtrauc zīdīšana.
Auglība
Pētījumi ar dzīvniekiem liecina, ka sorafenibs var pasliktināt vīriešu un sieviešu auglību (skatīt apakšpunktu 5.3).
04.7 Ietekme uz spēju vadīt transportlīdzekļus un apkalpot mehānismus
Nav veikti pētījumi par spēju vadīt transportlīdzekļus vai apkalpot mehānismus. Nav pamata uzskatīt, ka sorafenibs ietekmē spēju vadīt transportlīdzekļus vai apkalpot mehānismus.
04.8 Nevēlamās blakusparādības
Vissvarīgākās nopietnās blakusparādības bija miokarda išēmija un infarkts, kuņģa -zarnu trakta perforācija, zāļu hepatīts, asiņošana un hipertensija vai hipertensīva krīze.
Visbiežāk novērotās blakusparādības bija caureja, astēnija, alopēcija, infekcija, roku un kāju ādas reakcija (MedDRA atbilst "plaukstas-plantāra eritrodizestēzijas sindromam") un izsitumi.
Blakusparādības, par kurām ziņots dažādos klīniskos pētījumos vai pēcreģistrācijas periodā, ir uzskaitītas 1. tabulā, sakārtotas pēc MedDRA un biežuma. Biežums ir definēts šādi: ļoti bieži (≥1 / 10), bieži (≥1 / 100,
Katrā sastopamības biežumā nevēlamās blakusparādības ir norādītas dilstošā smaguma secībā.
1. tabula. Kopējās blakusparādības, par kurām ziņots pacientiem dažādos klīniskos pētījumos vai pēcreģistrācijas periodā.
* Nevēlamās reakcijas var būt dzīvībai bīstamas vai letālas. Šie notikumi ir retāk vai retāk nekā retāk.
** Roku un pēdu ādas reakcija atbilst plaukstas-plantāra eritrodizestēzijas sindromam MedDRA
Uzziniet vairāk par dažām blakusparādībām
Sastrēguma sirds mazspēja
uzņēmuma sponsorētā klīniskajā pētījumā sastrēguma sirds mazspēja tika ziņota par nevēlamu notikumu 1,9% pacientu, kuri tika ārstēti ar sorafenibu (N = 2276). Pētījumā 11213 (RCC) tika konstatētas nevēlamas blakusparādības, kas atbilst sastrēguma sirds mazspējai 1,7% ar sorafenibu ārstēto pacientu un 0,7% ar placebo ārstēto pacientu. Pētījumā 100554 (HCC) šādi gadījumi tika ziņoti 0,99% ar sorafenibu ārstēto pacientu un 1,1% ar placebo ārstēto pacientu.
Papildu informācija īpašām populācijām
Klīniskajos pētījumos noteiktas nevēlamas zāļu reakcijas, piemēram, roku un kāju ādas reakcija, caureja, alopēcija, svara zudums, hipertensija, hipokalciēmija un keratoakantoma / plakanšūnu ādas karcinoma, pacientiem ar diferencētu vairogdziedzera vēzi radās ievērojami biežāk nekā pacientiem. iekļauts nieru vai aknu šūnu karcinomas pētījumos.
Izmaiņas laboratorijas testos pacientiem ar HCC (3. pētījums) un RCC (1. pētījums)
Ļoti bieži ziņots par lipāzes un amilāzes līmeņa paaugstināšanos. 3. vai 4. pakāpes lipāzes līmeņa paaugstināšanās Kopējie toksicitātes kritērijiNevēlamie notikumi (CTCAE) radās attiecīgi 11% un 9% pacientu sorafeniba grupā 1. pētījumā (RCC) un 3. pētījumā (HCC), salīdzinot ar 7% un 9% pacientu Sorafenib grupā. CTCAE 3. pakāpe vai 4 amilāzes līmeņa paaugstināšanās novērota attiecīgi 1% un 2% pacientu sorafeniba grupā 1. un 3. pētījumā, salīdzinot ar 3% pacientu abās grupās.Par klīnisko pankreatītu ziņots 2 no 451 pacientam, kas tika ārstēti ar sorafenibu (CTCAE 4. pakāpe) 1. pētījumā, 1 no 297 pacientiem, kuri tika ārstēti ar sorafenibu (CTCAE 2. pakāpe) 3. pētījumā, un 1 no 451 pacientam (CTCAE 2. pakāpe). ar placebo 1. pētījumā.
Hipofosfatēmija ir ļoti izplatīts laboratorijas atklājums, un to novēroja attiecīgi 45% un 35% ar sorafenibu ārstēto pacientu 1. un 3. pētījumā, salīdzinot ar attiecīgi 12% un 11% ar placebo ārstētiem pacientiem. CTCAE 3. pakāpes hipofosfatēmija (1-2 mg / dl) 1. pētījumā novērota 13% pacientu, kas ārstēti ar sorafenibu, un 3% ar placebo ārstētiem pacientiem, savukārt 3. pētījumā-11% ar sorafenibu ārstētu pacientu un 2% pacientu, kas ārstēti ar placebo Nav ziņots par CTCAE 4. pakāpes hipofosfatēmijas gadījumiem (ar sorafenibu saistītās hipofosfatēmijas etioloģija nav zināma).
CTCAE 3. vai 4. pakāpes laboratorijas novirzes, tai skaitā limfopēnija un neitropēnija, tika novērotas ≥ 5% pacientu, kuri tika ārstēti ar sorafenibu.
Hipokalciēmija tika novērota 12% un 26,5% ar sorafenibu ārstēto pacientu, salīdzinot ar 7,5% un 14,8% pacientu placebo grupā attiecīgi 1. un 3. pētījumā. Hipokalciēmijas gadījumi bija viegli (CTCAE 1. un 2. pakāpe). CTCAE 3. pakāpes hipokalciēmija (6,0 - 7,0 mg / dl) radās 1,1% un 1,8% pacientu, kuri tika ārstēti ar sorafenibu, un 0,2% un 1,1% pacientu placebo grupā, un CTCAE 4. pakāpes hipokalciēmija (
1. un 3. pētījumā kālija samazināšanās tika novērota attiecīgi 5,4% un 9,5% pacientu, kuri tika ārstēti ar sorafenibu, salīdzinot ar 0,7% un 5,9% pacientu, kuri saņēma placebo. Lielākā daļa hipokaliēmijas gadījumu bija viegli (CTCAE 1. pakāpe). Šajos pētījumos CTCAE 3. pakāpes hipokaliēmija radās 1,1% un 0,4% ar sorafenibu ārstētiem pacientiem un 0,2% un 0,7% pacientu placebo grupā.
Izmaiņas laboratorijas testos pacientiem ar DTC (5. pētījums)
Hipokalciēmija tika novērota 35,7% pacientu, kuri tika ārstēti ar sorafenibu, salīdzinot ar 11,0% pacientu placebo grupā. Lielākā daļa hipokalciēmijas gadījumu bija vieglas. CTCAE 3. pakāpes hipokalciēmija radās 6,8% ar sorafenibu ārstēto pacientu un 1,9% pacientu placebo grupā, savukārt CTCAE 4. pakāpes hipokalciēmija radās 3,4% pacientu, kuri tika ārstēti ar sorafenibu, un 1,0% pacientu placebo grupā.
Citas klīniski nozīmīgas laboratorijas izmaiņas, kas novērotas 5. pētījumā, ir parādītas 2. tabulā.
2. tabula. Ārstēšanas izraisītas laboratorijas novirzes, par kurām ziņots pacientiem ar DTC (5. pētījums) dubultmaskētā fāzē
* Kopējie nelabvēlīgo notikumu terminoloģijas kritēriji (CTCAE), versija 3.0
** Ar sorafenibu saistītās hipofosfatēmijas etioloģija nav zināma.
Ziņošana par iespējamām blakusparādībām
Ir svarīgi ziņot par iespējamām blakusparādībām, kas rodas pēc zāļu reģistrācijas, jo tas ļauj nepārtraukti uzraudzīt zāļu ieguvuma / riska attiecību. Veselības aprūpes speciālistus lūdz ziņot par visām iespējamām blakusparādībām, izmantojot Itālijas Zāļu aģentūru, tīmekļa vietni : www.agenziafarmaco.gov.it/it/responsabili.
04.9 Pārdozēšana
Sorafeniba pārdozēšanas gadījumā nav īpašu ārstēšanas metožu. Lielākā klīniski pētītā sorafeniba deva ir 800 mg divas reizes dienā. Pēc šīs devas novērotās blakusparādības galvenokārt bija caureja un dermatoloģiskas reakcijas. Ja ir aizdomas par pārdozēšanu, sorafeniba lietošana jāpārtrauc un, ja nepieciešams, jāsāk atbalstoša terapija.
05.0 FARMAKOLOĢISKĀS ĪPAŠĪBAS
05.1 Farmakodinamiskās īpašības
Farmakoterapeitiskā grupa: pretaudzēju līdzekļi, proteīnkināzes inhibitori.
ATĶ kods: L01XE05.
Sorafenibs ir kināzes inhibitors, kam ir gan antiproliferatīvas, gan antiangiogēnas īpašības in vitro un in vivo.
Darbības mehānisms un farmakodinamiskā iedarbība
Sorafenibs ir kināzes inhibitors, kas kavē vēža šūnu proliferāciju in vitro. Sorafenibs kavē plaša spektra cilvēka audzēju augšanu, kas pārstādīti pelēm, kurām nav atopija, kā arī samazina audzēja angioģenēzi. Sorafenibs kavē audzēja šūnā esošo mērķu (CRAF, BRAF, V600E BRAF, c-KIT un FLT-) darbību. 3) un audzēja asinsvados (CRAF, VEGFR-2, VEGFR-3 un PDGFR-Ÿ). RAF kināzes ir serīna / treonīna kināzes, savukārt c-KIT, FLT-3, VEGFR-2, VEGFR-3 un PDGFR-Ã ir receptoru tirozīnkināzes.
Klīniskā efektivitāte
Sorafeniba drošība un klīniskā efektivitāte ir pētīta pacientiem ar aknu šūnu karcinomu (aknu šūnu karcinoma(HCC) pacientiem ar progresējošu nieru šūnu karcinomu (nieru šūnu karcinoma(RCC) un pacientiem ar diferencētu vairogdziedzera vēzi (diferencēta vairogdziedzera karcinoma, DTC).
Hepatokarcinoma
3. pētījums (pētījums 100554) bija daudzcentru, randomizēts, dubultmaskēts, placebo kontrolēts, starptautisks III fāzes klīniskais pētījums, kurā piedalījās 602 pacienti ar aknu šūnu vēzi. Sākotnējie demogrāfiskie rādītāji un slimības pazīmes bija salīdzināmas sorafeniba un placebo grupās attiecībā uz Estern Cooperative Oncology Group (ECOG) klasifikāciju (0: 54% pret 54%; 1. Pakāpe: 38% pret 39%; 2. Pakāpe: 8% pret 7%), TNM klasifikācijai (I posms:
Pētījums tika slēgts pēc tam, kad plānotā pagaidu vispārējās dzīvildzes (OS) analīze pārsniedza iepriekš noteikto efektivitātes robežu. Šī OS analīze parādīja statistiski nozīmīgu OS pieaugumu pacientiem, kas ārstēti ar sorafenibu, salīdzinot ar placebo ārstētiem pacientiem (HR: 0,69, p = 0,00058, skatīt 3. tabulu).
Šajā pētījumā dati par pacientiem ar Child Pugh B aknu darbības traucējumiem ir ierobežoti, un tika iekļauts tikai viens Child Pugh C pacients.
3. tabula. Efektivitātes rezultāti no 3. pētījuma (pētījums 100554) ar hepatokarcinomu
CI = ticamības intervāls, HR = riska attiecība (sorafenibs salīdzinājumā ar placebo)
* statistiski nozīmīgs, jo p vērtība bija zem noklusējuma O "robežvērtības, kas noteikta 0,0077
** neatkarīgs radioloģiskais pārskats
Otrajā III fāzes starptautiskajā, daudzcentru, randomizētā, dubultmaskētā, placebo kontrolētā pētījumā (4., 11849. pētījums) tika novērtēts sorafeniba klīniskais ieguvums 226 pacientiem ar progresējošu aknu vēzi. Šis pētījums, kas veikts Ķīnā, Korejā un Taivānā, apstiprināja 3. pētījuma rezultātus attiecībā uz sorafeniba labvēlīgo ieguvuma un riska profilu (HR (OS): 0,68, p = 0,01414).
Iepriekš definētajos stratifikācijas faktoros (ECOG klasifikācija, makroskopiskas asinsvadu invāzijas esamība vai neesamība un / vai audzēja ekstrahepatiska izplatība) 3. un 4. pētījumā HR pastāvīgi atbalstīja sorafenibu salīdzinājumā ar placebo. Izpētes apakšgrupu analīzes liecināja par mazāk izteiktu ārstēšanas efektu pacientiem ar tālu metastāzēm jau sākumā.
Nieru šūnu karcinoma
Sorafeniba panesamība un efektivitāte progresējošas nieru šūnu karcinomas (RCC) ārstēšanā tika pētīta divos klīniskajos pētījumos:
1. pētījums (11213 pētījums) bija daudzcentru, randomizēts, dubultmaskēts, placebo kontrolēts III fāzes klīniskais pētījums, kurā piedalījās 903 pacienti. Tika iekļauti tikai pacienti ar skaidru šūnu nieru audzējiem un ar zemu un vidēju riska faktoru saskaņā ar MSKCC. The galapunktsprimārā bija kopējā dzīvildze (OS, kopumā izdzīvošana) un izdzīvošana bez progresēšanas (PFS, Progresēšana Bezmaksas Izdzīvošana).
Apmēram pusei pacientu vispārējais stāvoklis bija vienāds ar 0 ECOG skalā, un puse pacientu piederēja prognostiskajai grupai ar zemu punktu skaitu saskaņā ar MSKCC klasifikāciju.
PFS tika novērtēts saskaņā ar RECIST kritērijiem ar aklu neatkarīgu radioloģisko pārskatu. PFS analīze tika veikta 342 gadījumos 769 pacientiem. Vidējais PFS vērtība bija 167 dienas ar sorafenibu ārstētiem pacientiem, salīdzinot ar 84 dienām pacientiem, kuri saņēma placebo (HR = 0,44; 95% TI: 0,35 - 0,55; p
"Analīze pagaidu (otrā analīze pagaidu) kopējai izdzīvošanai (kopumā izdzīvošana) tika veikta ar 367 nāves gadījumiem 903 pacientiem. Šīs analīzes nominālā alfa vērtība bija 0,0094. Ar sorafenibu ārstētiem pacientiem vidējā dzīvildze bija 19,3 mēneši, salīdzinot ar 15,9 mēnešiem pacientiem, kuri tika randomizēti placebo (HR = 0,77; 95% TI: 0,63-0,95; p = 0,015). Analīzes laikā aptuveni 200 pacienti pārgāja no placebo grupas uz sorafeniba grupu.
2. pētījums bija II fāzes pētījums ar nejaušinātu ārstēšanas pārtraukšanu pacientiem ar metastātisku vēzi, ieskaitot RCC. Pacienti ar stabilu slimību un sorafeniba terapiju tika randomizēti placebo vai sorafeniba terapijas turpināšanai. PFS pacientiem ar RCC bija ievērojami lielāks (163 dienas) ar sorafenibu ārstētiem pacientiem nekā tas, kas novērots pacientiem, kuri saņēma placebo (41 diena) (p = 0,0001, HR = 0,29).
Diferencēta vairogdziedzera karcinoma (DTC)
5. pētījums (14295. Pētījums) bija starptautisks, daudzcentru, randomizēts, dubultmaskēts, placebo kontrolēts III fāzes pētījums, kurā piedalījās 417 pacienti ar lokāli progresējošu vai metastātisku radioaktīvo jodu ugunsizturīgo DTC. Izdzīvošana bez slimības progresēšanas (PFS), ko noteica ar aklu neatkarīgu radioloģisku novērtējumu, pamatojoties uz RECIST kritērijiem, bija pētījuma primārais mērķa kritērijs. Sekundārie parametri ietvēra kopējo dzīvildzi (OS), audzēja atbildes reakciju un reakcijas ilgumu Pēc progresēšanas pacienti varēja saņemt atklātu sorafenibu.
Pacienti tika iekļauti pētījumā, ja tie progresēja 14 mēnešu laikā pirms uzņemšanas un ja viņiem bija DTC, kas nav izturīgs pret radiojodu (radioaktīvais jods, RAI). RAI rezistenti DTC tika definēti kā ne-jodu pastiprinoša bojājuma klātbūtne RAI scintigrāfijā vai RAI kumulatīva ievadīšana ≥ 22,2 GBq vai progresēšana pēc RAI terapijas iepriekšējo 16 mēnešu laikā. ne vairāk kā 16 mēnešu attālumā viens no otra.
Sākotnējā demogrāfija un pacientu īpašības abās ārstēšanas grupās bija labi līdzsvarotas. Metastāzes bija plaušās 86%, limfmezglos 51% un kaulos 27% pacientu. Vidējā kumulatīvā radiojoda aktivitāte, kas tika ievadīta pirms reģistrācijas, bija aptuveni 14,8 GBq. Lielākajai daļai pacientu bija papilārā karcinoma (56,8%), kam sekoja folikulāra karcinoma (25,4%) un vāji diferencēta karcinoma (9,6%).
Vidējais laiks līdz PFS bija 10,8 mēneši sorafeniba grupā, salīdzinot ar 5,8 mēnešiem placebo grupā. (HR = 0,587; 95% ticamības intervāls (TI): 0,454, 0,758; p vienpusējs
Sorafeniba ietekme uz PFS bija nemainīga neatkarīgi no ģeogrāfiskā reģiona, vecuma virs vai zem 60 gadiem, dzimuma, histoloģijas un kaulu metastāžu klātbūtnes vai neesamības.
Vispārējās dzīvildzes analīzē, kas tika veikta 9 mēnešus pēc beigu PFS analīzes beigu datuma, statistiski nozīmīgas atšķirības starp kopējo dzīvildzi starp ārstēšanas grupām nebija (HR 0,884; TI 95 %: 0,633; 1,236, vienpusēja p- vērtība ir 0,236). Vidējā OS netika sasniegta sorafeniba grupā, bet placebo grupā tas bija 36,5 mēneši. Simt piecdesmit septiņi pacienti (75%), kuri tika randomizēti placebo grupā, un 61 pacients (30%), kuri tika randomizēti sorafeniba grupā, saņēma atklātu sorafenibu.
Vidējais terapijas ilgums dubultmaskētā fāzē bija 46 nedēļas (diapazons 0,3-135) pacientiem, kuri saņēma sorafenibu, un 28 nedēļas (diapazons 1,7-132) pacientiem, kuri saņēma placebo.
Netika novērota pilnīga reakcija (pilnīga atbilde, CR) saskaņā ar RECIST kritērijiem. Kopējais reakcijas līmenis (CR + daļēja reakcija, daļēja reakcija (PR)), ko noteica neatkarīgs radioloģisks novērtējums, bija augstāks sorafeniba grupā (24 pacienti, 12,2%) salīdzinājumā ar placebo grupu (1 pacients, 0,5%), p vienpusējs
Apakšgrupu pēc-analīze, pamatojoties uz maksimālo audzēja lielumu, parādīja ārstēšanas ietekmi uz PFS par labu sorafenibam, salīdzinot ar placebo, pacientiem ar maksimālo audzēja bojājuma izmēru 1,5 cm vai vairāk (HR 0,54 (95% TI: 0,41-0,71)) , savukārt skaitliski mazāks efekts tika reģistrēts pacientiem ar maksimālo audzēja bojājuma izmēru, kas mazāks par 1,5 cm (HR 0,87 (95% TI: 0,40 -1,89)).
Post-hoc analīze, kuras pamatā bija sākotnējie stāvokļi, kas saistīti ar vairogdziedzera vēža simptomiem, parādīja ārstēšanas ietekmi uz PFS par labu sorafenibam, salīdzinot ar placebo, gan simptomātiskiem, gan asimptomātiskiem pacientiem. HR vērtība brīvās dzīvildzes progresēšanai bija 0,39 (95% TI: 0,21- 0,72) pacientiem ar simptomiem sākotnēji un 0,60 (95% TI: 0,45 - 0,81) pacientiem bez simptomiem pamata apstākļos.
QT intervāla pagarināšana
Klīniskās farmakoloģijas pētījumā QT / QTc tika mērīts 31 pacientam sākumā (pirms ārstēšanas) un pēc ārstēšanas. Pēc 28 dienu ārstēšanas cikla maksimālās sorafeniba koncentrācijas laikā QTcB pagarinājās par 4 ± 19 ms un QTcF par 9 ± 18 ms, salīdzinot ar placebo grupas sākotnējo stāvokli. EKG uzraudzības laikā pēc ārstēšanas nevienam pacientam QTcB vai QTcF vērtība nepārsniedza 500 ms (skatīt apakšpunktu 4.4).
Pediatriskā populācija
Eiropas Zāļu aģentūra ir atcēlusi pienākumu iesniegt pētījumu rezultātus visās pediatriskās populācijas apakšgrupās attiecībā uz nieru un nieru iegurņa vēzi (izņemot nefroblastomu, nefroblastomatozi, skaidru šūnu sarkomu, mezoblastisko nefromu, nieru medulāro karcinomu un nieru rabdoīdu audzēju). un aknu karcinoma un intrahepatiskais žultsvads (izņemot hepatoblastomu) un diferencēta vairogdziedzera karcinoma (informāciju par lietošanu bērniem skatīt 4.2. apakšpunktā).
05.2 "Farmakokinētiskās īpašības
Absorbcija un izplatība
Pēc sorafeniba tablešu lietošanas vidējā relatīvā biopieejamība ir 38–49%, salīdzinot ar šķīdumu iekšķīgai lietošanai. Absolūtā biopieejamība nav zināma. Pēc iekšķīgas lietošanas sorafenibs sasniedz maksimālo koncentrāciju plazmā aptuveni 3 stundu laikā. Lietojot maltīti ar augstu tauku saturu, sorafeniba uzsūkšanās samazinās par aptuveni 30%, salīdzinot ar lietošanu tukšā dūšā.
Vidējās Cmax un AUC palielinās mazāk nekā proporcionāli, lietojot devas virs 400 mg divas reizes dienā. Sorafeniba saistīšanās ar plazmas olbaltumvielām in vitro ir 99,5%.
Atkārtota sorafeniba lietošana 7 dienas izraisīja 2,5 līdz 7 reizes lielāku uzkrāšanos, salīdzinot ar vienreizēju lietošanu. Sorafeniba līdzsvara stāvoklis tiek sasniegts 7 dienu laikā, vidējās maksimālās un minimālās koncentrācijas plazmā attiecība ir mazāka par 2.
Pacientiem ar DTC, RCC un HCC tika noteikta sorafeniba līdzsvara koncentrācija, lietojot 400 mg divas reizes dienā. Vislielākā vidējā koncentrācija tika novērota pacientiem ar DTC (aptuveni divreiz lielāka nekā pacientiem ar RCC un HCC), bet mainīgums bija liels visi audzēju veidi Šīs augstākās koncentrācijas cēlonis pacientiem ar DTC nav zināms.
Biotransformācija un eliminācija
Sorafeniba eliminācijas pusperiods ir aptuveni 25 līdz 48 stundas. Sorafenibs galvenokārt tiek metabolizēts aknās, izmantojot CYP3A4 mediētu oksidatīvo metabolismu un UGT1A9 mediēto glikurono konjugāciju. Konjugēts sorafenibs var izdalīties kuņģa -zarnu traktā ar dažu baktēriju glikuronidāzes aktivitāti, tādējādi ļaujot nekonjugētās aktīvās sastāvdaļas reabsorbcijai. Ir novērots, ka kombinācija ar neomicīnu traucē šo procesu, samazinot sorafeniba vidējo biopieejamību par 54%.
Sorafenibs veido aptuveni 70–85% analītu, kas cirkulē līdzsvara plazmā. Ir identificēti astoņi sorafeniba metabolīti, no kuriem pieci ir atrasti plazmā. Galvenajam sorafeniba metabolītam, kas cirkulē plazmā, ir piridīna N-oksīdsin vitro līdzīgi kā sorafenibam. Šis metabolīts veido aptuveni 9–16% analītisko vielu, kas cirkulē līdzsvara stāvoklī.
Pēc 100 mg sorafeniba šķīduma devas iekšķīgas lietošanas 96% devas tika atgūti 14 dienu laikā: 77% izkārnījumos un 19% urīnā kā glikuronāta metabolīti. Neizmainīts sorafenibs, kas veido 51% no devas, tika konstatēts izkārnījumos, bet ne urīnā, norādot, ka nemetabolizētās aktīvās vielas izdalīšanās ar žulti var veicināt sorafeniba elimināciju.
Farmakokinētika noteiktās pacientu kategorijās
Demogrāfisko datu analīze parādīja, ka nav korelācijas starp farmakokinētiku un vecumu (līdz 65 gadiem), dzimumu vai ķermeņa svaru.
Pediatriskā populācija
Nav veikti pētījumi, lai pārbaudītu sorafeniba farmakokinētiku bērniem.
Rase
Klīniski nozīmīgas farmakokinētikas atšķirības starp baltās rases un Āzijas indivīdiem nepastāv.
Nieru darbības traucējumi
Četros I fāzes klīniskajos pētījumos sorafeniba ekspozīcija līdzsvara stāvoklī pacientiem ar viegliem vai vidēji smagiem nieru darbības traucējumiem bija līdzīga tai, kas konstatēta pacientiem ar normālu nieru darbību. Klīniskā farmakoloģijas pētījumā (400 mg vienreizēja sorafeniba deva) netika novērota saistība starp sorafeniba iedarbība un nieru darbība pacientiem ar normālu nieru darbību vai viegliem, vidēji smagiem vai smagiem nieru darbības traucējumiem. Nav pieejami dati par pacientiem, kuriem nepieciešama dialīze.
Aknu darbības traucējumi
Pacientiem ar hepatocelulāru karcinomu (HCC) un ar aknu darbības traucējumiem, kas novērtēti kā A vai B Child-Pugh (viegls vai vidēji smags), iedarbības vērtības bija salīdzināmas un bija robežās, kas novērotas pacientiem bez aknu darbības traucējumiem. Sorafeniba farmakokinētika A un B pakāpes Child-Pugh pacientiem bez HCC bija līdzīga veseliem brīvprātīgajiem. Nav datu par pacientiem ar smagiem (Child-Pugh C) aknu darbības traucējumiem. Sorafenibs tiek izvadīts galvenokārt caur aknām, un iedarbība šajā pacientu grupā var palielināties.
05.3 Preklīniskie drošības dati
Sorafeniba preklīniskais drošības profils tika novērtēts pelēm, žurkām, suņiem un trušiem.
Atkārtotu devu toksicitātes pētījumi atklāja izmaiņas dažādos orgānos (deģenerācija un reģenerācija), ja iedarbība bija zemāka par klīniskajos pētījumos izmantoto devu iedarbību (pamatojoties uz AUC salīdzinājumu).
Pēc atkārtotas dozēšanas jauniem un augošiem suņiem iedarbība uz kauliem un zobiem tika novērota, ja iedarbība bija zemāka par klīniskajos pētījumos izmantoto devu iedarbību. Šīs sekas bija nevienmērīga augšstilba kaula augšanas plāksnes sabiezēšana, medulāra hipoplāzija izmainīto augšanas plākšņu tuvumā un izmaiņas dentīna sastāvā. Pieaugušam sunim līdzīga iedarbība netika izraisīta.
Tika veikta standarta genotoksicitātes pētījumu programma, un tika iegūti pozitīvi rezultāti, jo vienā testā tika konstatēts hromosomu strukturālo aberāciju pieaugums. in vitro zīdītāju šūnās (ķīniešu kāmju olnīcās) klastogenitātes noteikšanai vielmaiņas aktivācijas klātbūtnē. Sorafenibs nebija genotoksisks Ames testā vai mikrokodolu testā in vivo pelē. Ražošanas procesa starpprodukts, kas ir arī gala aktīvajā vielā (in vitro uz baktēriju šūnām (Ames tests). Turklāt sorafeniba partija, kas pārbaudīta ar standarta genotoksisku bateriju, ietvēra 0,34% PAPE.
Kancerogenitātes pētījumi ar sorafenibu nav veikti.
Īpaši pētījumi ar dzīvniekiem ar sorafenibu nav veikti, lai novērtētu ietekmi uz auglību. Tomēr ir sagaidāma nelabvēlīga ietekme uz vīriešu un sieviešu auglību, jo atkārtotu devu pētījumi ar dzīvniekiem parādīja izmaiņas vīriešu un sieviešu reproduktīvajos orgānos, ja iedarbība bija zemāka par klīniskajos pētījumos izmantoto devu iedarbību (pamatojoties uz AUC). Izmaiņas parasti sastāvēja no žurku deģenerācijas pazīmes un sēklinieku, epididimijas, prostatas un sēklas pūslīšu attīstības aizkavēšanās. Žurku mātītēm bija dzelteno ķermeņa centrālā nekroze un olnīcu folikulu attīstības aizsprostojums. Suņiem olnīcās bija cauruļveida deģenerācija. sēklinieki un oligospermija.
Ir pierādīts, ka sorafenibs ir embriotoksisks un teratogēns, ja to lieto žurkām un trušiem, ja iedarbība ir zemāka par klīniskajos pētījumos izmantotajām devām. Novērotā ietekme ietvēra mātes un augļa ķermeņa masas samazināšanos, augļa rezorbciju skaita palielināšanos un palielinātu ārējo un viscerālo anomāliju skaitu.
Vides riska novērtējuma pētījumi parādīja, ka sorafeniba tozilāts ir potenciāli noturīgs, bioakumulatīvs un toksisks videi. Informācija par vides riska novērtējumu ir pieejama šo zāļu Eiropas publiskā novērtējuma ziņojumā (EPAR) (skatīt 6.6. Apakšpunktu).
06.0 FARMACEITISKĀ INFORMĀCIJA
06.1 Palīgvielas
Planšetdatora kodols:
Nātrija kroskarmeloze
Mikrokristāliskā celuloze
Hipromeloze
Nātrija laurilsulfāts
Magnija stearāts
Tabletes pārklājums:
Hipromeloze
Makrogols
Titāna dioksīds (E 171)
Sarkanais dzelzs oksīds (E 172)
06.2 Nesaderība
Nav būtisks.
06.3 Derīguma termiņš
3 gadi.
06.4 Īpaši uzglabāšanas nosacījumi
Uzglabāt temperatūrā līdz 25 ° C.
06.5 Tiešā iepakojuma veids un iepakojuma saturs
Kastīte ar 112 apvalkotām tabletēm (4 x 28) caurspīdīgā blisterī (PP / alumīnijs).
06.6 Norādījumi lietošanai un lietošanai
Šīs zāles var radīt potenciālu risku videi. Neizlietotās zāles un šo zāļu atkritumi jāiznīcina saskaņā ar vietējiem noteikumiem.
07.0 REĢISTRĀCIJAS APLIECĪBAS ĪPAŠNIEKS
Bayer Pharma AG
13342 Berlīne
Vācija
08.0 REĢISTRĀCIJAS APLIECĪBAS NUMURS
EU/1/06/342/001
037154010
09.0 PIRMĀJAS APLIECĪBAS VAI ATĻAUJAS DATUMS
Reģistrācijas datums: 2006. gada 19. jūlijs
Pēdējās pārreģistrācijas datums: 2011. gada 21. jūlijs
10.0 TEKSTA PĀRSKATĪŠANAS DATUMS
05/2014